Analyses of Tissue Culture Adaptation of Human Herpesvirus-6A by Whole Genome Deep Sequencing Redefines the Reference Sequence and Identifies Virus Entry Complex Changes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Illumina Deep Sequencing
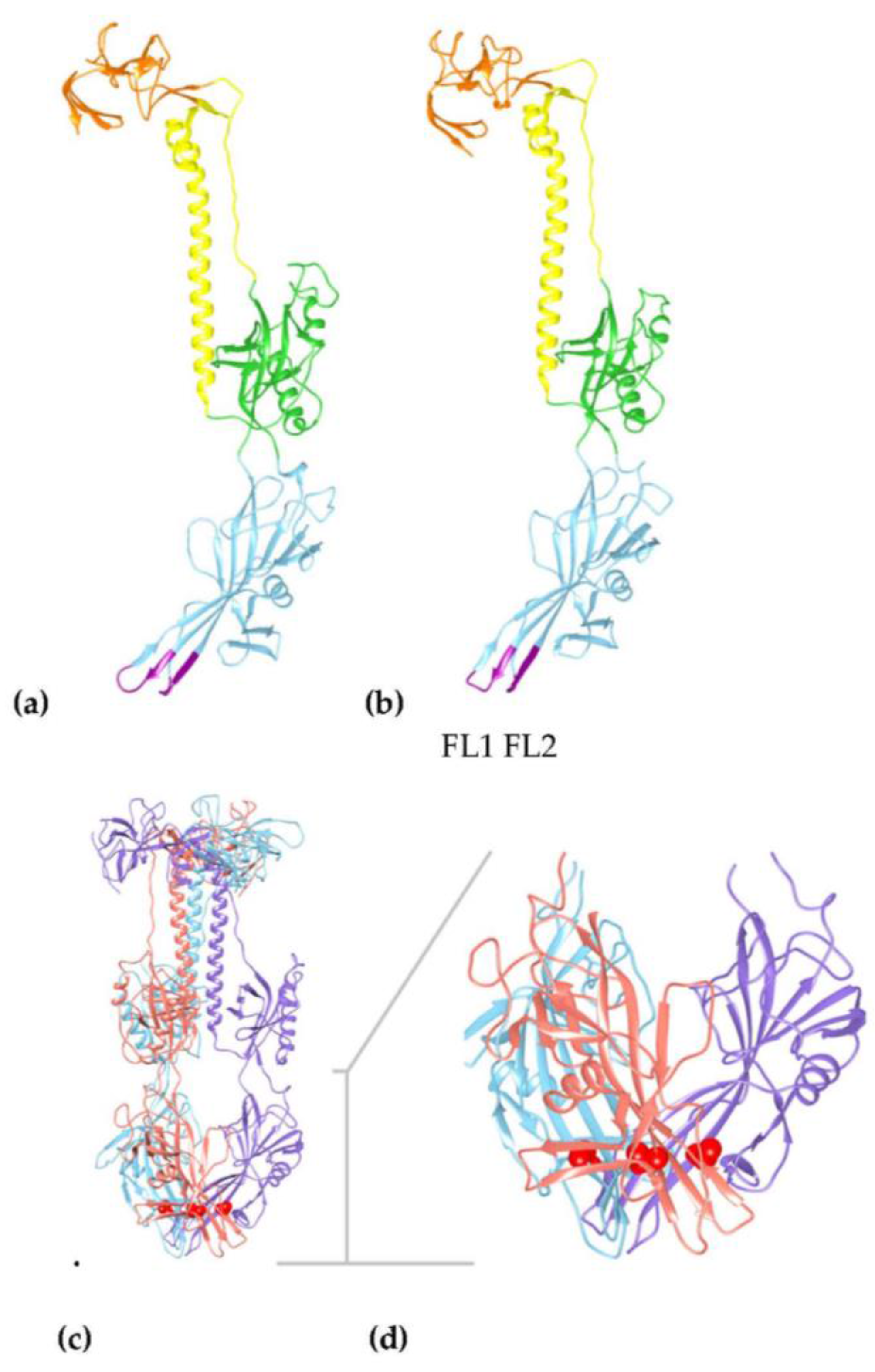
2.3. Molecular Models
3. Results
3.1. Deep Sequence Analyses of Reference HHV-6A Strain U1102 Shows Corrections
3.2. Changes with BACmid Cloning and Reconstruction
3.3. Emerging Variant Genome Populations Generated after Tissue Culture Passage
3.4. Virus Envelope Genes Mutations from Tissue Culture Passage Affect Fusion Complex
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A., 3rd; Kouzarides, T.; Martignetti, J.A.; et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar] [PubMed]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [PubMed]
- Ciferri, C.; Chandramouli, S.; Donnarumma, D.; Nikitin, P.A.; Cianfrocco, M.A.; Gerrein, R.; Feire, A.L.; Barnett, S.W.; Lilja, A.E.; Rappuoli, R.; et al. Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Murrell, I.; Wilkie, G.S.; Davison, A.J.; Statkute, E.; Fielding, C.A.; Tomasec, P.; Wilkinson, G.W.; Stanton, R.J. Genetic Stability of Bacterial Artificial Chromosome-Derived Human Cytomegalovirus during Culture In Vitro. J. Virol. 2016, 90, 3929–3943. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Chandramouli, S.; Leitner, A.; Donnarumma, D.; Cianfrocco, M.A.; Gerrein, R.; Friedrich, K.; Aggarwal, Y.; Palladino, G.; Aebersold, R.; et al. Antigenic Characterization of the HCMV gH/gL/gO and Pentamer Cell Entry Complexes Reveals Binding Sites for Potently Neutralizing Human Antibodies. PLoS Pathog. 2015, 11, e1005230. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Sacha, J.B.; Hughes, C.M.; Ford, J.C.; Burwitz, B.J.; Scholz, I.; Gilbride, R.M.; Lewis, M.S.; Gilliam, A.N.; Ventura, A.B.; et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 2013, 340, 1237874. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.A.; Dyer, A.P.; Milne, R.S.; Sevilla-Reyes, E.; Gompels, U.A. A role for human cytomegalovirus glycoprotein O (gO) in cell fusion and a new hypervariable locus. Virology 2002, 293, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.A.; Gompels, U.A. N- and C-terminal external domains of human herpesvirus-6 glycoprotein H affect a fusion-associated conformation mediated by glycoprotein L binding the N terminus. J. Gen. Virol. 1999, 80, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.A.; Liu, D.X.; Gompels, U.A. Definition of a human herpesvirus-6 betaherpesvirus-specific domain in glycoprotein gH that governs interaction with glycoprotein gL: Substitution of human cytomegalovirus glycoproteins permits group-specific complex formation. Virology 1996, 217, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Jasirwan, C.; Furusawa, Y.; Tang, H.; Maeki, T.; Mori, Y. Human herpesvirus-6A gQ1 and gQ2 are critical for human CD46 usage. Microbiol. Immunol. 2014, 58, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Yang, X.; Akkapaiboon, P.; Okuno, T.; Yamanishi, K. Human herpesvirus 6 variant A glycoprotein H-glycoprotein L-glycoprotein Q complex associates with human CD46. J. Virol. 2003, 77, 4992–4999. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hayashi, M.; Maeki, T.; Yamanishi, K.; Mori, Y. Human herpesvirus 6 glycoprotein complex formation is required for folding and trafficking of the gH/gL/gQ1/gQ2 complex and its cellular receptor binding. J. Virol. 2011, 85, 11121–11130. [Google Scholar] [CrossRef] [PubMed]
- Dewin, D.R.; Catusse, J.; Gompels, U.A. Identification and characterization of U83A viral chemokine, a broad and potent beta-chemokine agonist for human CCRs with unique selectivity and inhibition by spliced isoform. J. Immunol. 2006, 176, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.; Spyrou, M.A.; Hubacek, P.; Kuhl, U.; Lassner, D.; Gompels, U.A. Analyses of germline, chromosomally integrated human herpesvirus 6A and B genomes indicate emergent infection and new inflammatory mediators. J. Gen. Virol. 2015, 96, 370–389. [Google Scholar] [CrossRef] [PubMed]
- French, C.; Menegazzi, P.; Nicholson, L.; Macaulay, H.; DiLuca, D.; Gompels, U.A. Novel, nonconsensus cellular splicing regulates expression of a gene encoding a chemokine-like protein that shows high variation and is specific for human herpesvirus 6. Virology 1999, 262, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Schleimann, M.H.; Hoberg, S.; Solhoj Hansen, A.; Bundgaard, B.; Witt, C.T.; Kofod-Olsen, E.; Hollsberg, P. The DR6 protein from human herpesvirus-6B induces p53-independent cell cycle arrest in G2/M. Virology 2014, 452–453, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, E.; Kouvatsis, V.; Dimitriadis, G.; Inoue, N.; Arsenakis, M. Identification and characterization of the gene products of open reading frame U86/87 of human herpesvirus 6. Virus Res. 2002, 89, 89–101. [Google Scholar] [CrossRef]
- Salahuddin, S.Z.; Ablashi, D.V.; Markham, P.D.; Josephs, S.F.; Sturzenegger, S.; Kaplan, M.; Halligan, G.; Biberfeld, P.; Wong-Staal, F.; Kramarsky, B.; et al. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 1986, 234, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.A. Human herpesvirus 6A, 6B and 7 (Herpesviridae). In Reference Module in Biomedical Sciences, Encyclopedia of Virology, 3rd ed.; Mahy, B., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 498–505. [Google Scholar]
- Simpson, K.E.; Storch, G.A.; Lee, C.K.; Ward, K.E.; Danon, S.; Simon, C.M.; Delaney, J.W.; Tong, A.; Canter, C.E. High Frequency of Detection by PCR of Viral Nucleic Acid in The Blood of Infants Presenting with Clinical Myocarditis. Pediatr. Cardiol. 2016, 37, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Stefanski, H.E.; Thibert, K.A.; Pritchett, J.; Prusty, B.K.; Wagner, J.E.; Lund, T.C. Fatal Myocarditis Associated With HHV-6 Following Immunosuppression in Two Children. Pediatrics 2016, 137. [Google Scholar] [CrossRef] [PubMed]
- Gravel, A.; Dubuc, I.; Morissette, G.; Sedlak, R.H.; Jerome, K.R.; Flamand, L. Inherited chromosomally integrated human herpesvirus 6 as a predisposing risk factor for the development of angina pectoris. Proc. Natl. Acad. Sci. USA 2015, 112, 8058–8063. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, U.; Lassner, D.; Wallaschek, N.; Gross, U.M.; Krueger, G.R.; Seeberg, B.; Kaufer, B.B.; Escher, F.; Poller, W.; Schultheiss, H.P. Chromosomally integrated human herpesvirus 6 in heart failure: Prevalence and treatment. Eur. J. Heart Fail. 2015, 17, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.; Spyrou, M.A.; Pearson, M.; Lassner, D.; Kuhl, U.; Gompels, U.A. Complete Genome Sequence of Germline Chromosomally Integrated Human Herpesvirus 6A and Analyses Integration Sites Define a New Human Endogenous Virus with Potential to Reactivate as an Emerging Infection. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Caserta, M.T.; Hall, C.B.; Canfield, R.L.; Davidson, P.; Lofthus, G.; Schnabel, K.; Carnahan, J.; Shelley, L.; Wang, H. Early developmental outcomes of children with congenital HHV-6 infection. Pediatrics 2014, 134, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.A.; Nicholas, J.; Lawrence, G.; Jones, M.; Thomson, B.J.; Martin, M.E.; Efstathiou, S.; Craxton, M.; Macaulay, H.A. The DNA sequence of human herpesvirus-6: Structure, coding content, and genome evolution. Virology 1995, 209, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Kawabata, A.; Yoshida, M.; Oyaizu, H.; Maeki, T.; Yamanishi, K.; Mori, Y. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology 2010, 407, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Gulve, N.; Kimmerling, K.; Johnston, A.D.; Krueger, G.R.; Ablashi, D.V.; Prusty, B.K. Anti-herpesviral effects of a novel broad range anti-microbial quaternary ammonium silane, K21. Antivir. Res. 2016, 131, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.G.; Prusty, B.K.; Gompels, U.A. Use of whole genome deep sequencing to define emerging minority variants in virus envelope genes in herpesvirus treated with novel antimicrobial K21. Antivir. Res. 2017, 146, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.D.; Dillon, G.P.; Degrave, W.S.; Berriman, M. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res. 2011, 39, e57. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.; Spyrou, M.A.; Donaldson, C.D.; Depledge, D.; Breuer, J.; Gompels, U.A. Complete Genome Sequence of the Human Herpesvirus 6A Strain AJ from Africa Resembles Strain GS from North America. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Ellinger, K.; Fleckenstein, B. Gene for the major antigenic structural protein (p100) of human herpesvirus 6. J. Virol. 1992, 66, 3918–3924. [Google Scholar] [PubMed]
- Nicholas, J.; Martin, M.E. Nucleotide sequence analysis of a 38.5-kilobase-pair region of the genome of human herpesvirus 6 encoding human cytomegalovirus immediate-early gene homologs and transactivating functions. J. Virol. 1994, 68, 597–610. [Google Scholar] [PubMed]
- Adler, H.; Messerle, M.; Wagner, M.; Koszinowski, U.H. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 2000, 74, 6964–6974. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Choudhury, S.; Kashanchi, F.; Doniger, J.; Berneman, Z.; Frenkel, N.; Rosenthal, L.J. A transforming fragment within the direct repeat region of human herpesvirus type 6 that transactivates HIV-1. Oncogene 1994, 9, 1167–1175. [Google Scholar] [PubMed]
- Kondo, K.; Shimada, K.; Sashihara, J.; Tanaka-Taya, K.; Yamanishi, K. Identification of human herpesvirus 6 latency-associated transcripts. J. Virol. 2002, 76, 4145–4151. [Google Scholar] [CrossRef] [PubMed]
- Akkapaiboon, P.; Mori, Y.; Sadaoka, T.; Yonemoto, S.; Yamanishi, K. Intracellular processing of human herpesvirus 6 glycoproteins Q1 and Q2 into tetrameric complexes expressed on the viral envelope. J. Virol. 2004, 78, 7969–7983. [Google Scholar] [CrossRef] [PubMed]
- Burke, H.G.; Heldwein, E.E. Crystal Structure of the Human Cytomegalovirus Glycoprotein B. PLoS Pathog. 2015, 11, e1005227. [Google Scholar] [CrossRef] [PubMed]
- Santoro, F.; Kennedy, P.E.; Locatelli, G.; Malnati, M.S.; Berger, E.A.; Lusso, P. CD46 is a cellular receptor for human herpesvirus 6. Cell 1999, 99, 817–827. [Google Scholar] [CrossRef]
- Hansen, A.S.; Bundgaard, B.B.; Biltoft, M.; Rossen, L.S.; Hollsberg, P. Divergent tropism of HHV-6AGS and HHV-6BPL1 in T cells expressing different CD46 isoform patterns. Virology 2017, 502, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Zeev-Ben-Mordehai, T.; Vasishtan, D.; Hernandez Duran, A.; Vollmer, B.; White, P.; Prasad Pandurangan, A.; Siebert, C.A.; Topf, M.; Grunewald, K. Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein B. Proc. Natl. Acad. Sci. USA 2016, 113, 4176–4181. [Google Scholar] [CrossRef] [PubMed]
- Achour, A.; Malet, I.; Le Gal, F.; Dehee, A.; Gautheret-Dejean, A.; Bonnafous, P.; Agut, H. Variability of gB and gH genes of human herpesvirus-6 among clinical specimens. J. Med. Virol. 2008, 80, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Franti, M.; Aubin, J.T.; Gautheret-Dejean, A.; Malet, I.; Cahour, A.; Huraux, J.M.; Agut, H. Preferential associations of alleles of three distinct genes argue for the existence of two prototype variants of human herpesvirus 7. J. Virol. 1999, 73, 9655–9658. [Google Scholar] [PubMed]
- Gravel, A.; Ablashi, D.; Flamand, L. Complete Genome Sequence of Early Passaged Human Herpesvirus 6A (GS Strain) Isolated from North America. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.E.; Thomson, B.J.; Honess, R.W.; Craxton, M.A.; Gompels, U.A.; Liu, M.Y.; Littler, E.; Arrand, J.R.; Teo, I.; Jones, M.D. The genome of human herpesvirus 6: Maps of unit-length and concatemeric genomes for nine restriction endonucleases. J. Gen. Virol. 1991, 72, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Gulve, N.; Frank, C.; Klepsch, M.; Prusty, B.K. Chromosomal integration of HHV-6A during non-productive viral infection. Sci. Rep. 2017, 7, 512. [Google Scholar] [CrossRef] [PubMed]
- Wallaschek, N.; Gravel, A.; Flamand, L.; Kaufer, B.B. The putative U94 integrase is dispensable for human herpesvirus 6 (HHV-6) chromosomal integration. J. Gen. Virol. 2016, 97, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Wallaschek, N.; Sanyal, A.; Pirzer, F.; Gravel, A.; Mori, Y.; Flamand, L.; Kaufer, B.B. The Telomeric Repeats of Human Herpesvirus 6A (HHV-6A) Are Required for Efficient Virus Integration. PLoS Pathog. 2016, 12, e1005666. [Google Scholar] [CrossRef] [PubMed]
- Ruedas, J.B.; Ladner, J.T.; Ettinger, C.R.; Gummuluru, S.; Palacios, G.; Connor, J.H. Spontaneous Mutation at Amino Acid 544 of the Ebola Virus Glycoprotein Potentiates Virus Entry and Selection in Tissue Culture. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.A.; Carss, A.L.; Saxby, C.; Hancock, D.C.; Forrester, A.; Minson, A.C. Characterization and sequence analyses of antibody-selected antigenic variants of herpes simplex virus show a conformationally complex epitope on glycoprotein H. J. Virol. 1991, 65, 2393–2401. [Google Scholar] [PubMed]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Massaro, R.; Campadelli-Fiume, G. Dissociation of HSV gL from gH by alphavbeta6- or alphavbeta8-integrin promotes gH activation and virus entry. Proc. Natl. Acad. Sci. USA 2015, 112, E3901–E3910. [Google Scholar] [CrossRef] [PubMed]
- Vallbracht, M.; Brun, D.; Tassinari, M.; Vaney, M.C.; Pehau-Arnaudet, G.; Guardado-Calvo, P.; Haouz, A.; Klupp, B.G.; Mettenleiter, T.C.; Rey, F.A.; et al. Structure-function dissection of the Pseudorabies virus glycoprotein B fusion loops. J. Virol. 2017. [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Amplicon | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Size (bp) |
|---|---|---|---|
| DR1 | ACAACCGCATCTTCTTCAG | ATGAACGGAGATCTGGGAG | 1750 |
| DR1DR6 | CCTCATCTGTTATTCCCTTCC | ACTCACCGCAGTCTTGAAC | 2872 |
| DR6 | TCCAAACTGTACGGCTATG | TGGCGTCTAATCCAGGTAC | 1858 |
| DR_UL | CATGAGAGGACACTGGACGTACTC | GACATCTGTGGACCATGCA | 2339 |
| 0203 | ACCATGTGCGGAGAAAGTTG | ACGAGAATGAATATGCCGATG | 2174 |
| 0306 | ACTCCTATCCGTCTAAACTGCT | ATTTCTCACGCCGGTATTC | 4417 |
| 0611 | ATGCCAAATCTAGCTGCTG | GAATCGGAGGAAGCTAACGT | 4757 |
| 1114 | TCTTCAGGTGTCTCAACCTTC | TGATGATCTGCTGTTGGAAC | 5737 |
| 1419 | TTGAGATGCAGAGCCTCTC | ACGTGGTGGATAATTCTTCAG | 5370 |
| 1923 | CTACATCATCGGTTGCCATA | TACAGAAGTTTCGGCAACTG | 5216 |
| 2328 | TAGTCGCTGGATGTCGATAC | GAGGAATTGTGGACAACGCT | 4721 |
| 2830 | CATCCAACTCCTTGTCCGT | TCGTTCGACGTGTCTGAC | 5753 |
| 3031 | GACACTGTCCAACGCATCT | CTCTTGGGCATAAGTCCAGTA | 6552 |
| 3134 | ACAGAGCGACACCCAATATTC | AGCTGTGTCGATGTCTTGCTA | 2787 |
| 3439 | TGCATTCTCTCACAGCATG | TGTTTCCTTATGCCACCAATC | 6309 |
| 3941 | TGAGGCATGAGACTCAGAAG | AGCGGCATCCATAATGCTTA | 4796 |
| 4142 | AGCATCGTTTGAAGACACC | GAGTTGTTGTTCGCTGGTCT | 2810 |
| 4142INT | TTGTTTCAGGCCAGCATC | ATACCGAAAACTCAGGCAAC | 2983 |
| 4245 | AGACCGCATTCGTATCGCT | CACGATCAAGTTCGAATCGTA | 5092 |
| 4549 | GAGTGCTCGGATAAGTTCATT | ACGATACAATCCGGAATGC | 5922 |
| 4953 | TGGAACTCGGAATTGTTTCT | GTTTGAGACTGTATCTGCGCA | 4282 |
| 5357 | TATGGTGAAGAACCGTCG | TCCGCAGATAACAGAATGG | 4707 |
| 5758 | CAGTGCCATCTGTTATGAATAG | AGTCGCAGTAAGGTCCACGT | 6792 |
| 5864 | TGCTGAAATTACCTCAGTGAG | CCATTCAGTATAATGCATCC | 4229 |
| 6469 | CTGTATCAATATCAAGGCGG | ACCACTGAATACGAACGCTC | 5111 |
| 6974 | GCGATTGTTCTGCGATAGAG | CTTATATCCGAGCCTTGCAGT | 6472 |
| 7476 | GAGAACATGCTAGACAATTGG | CAAGAACTGCGACTCAATCC | 2668 |
| 7679 | GTTGCTAGTTGTATGACTTGG | CAATATCCACCGTTAGAGAAC | 7681 |
| 7984 | AGATGTATGCTGAAGAACGTG | GGATCGTCAACCGTTAGTG | 5109 |
| 8486 | CACAGTGTGTTCGCCGGAAG | ACAGACAATGCACATCCTCTG | 4773 |
| 8690 | AGGTTGATACGGCAACGAG | GCAATCATTAGCATACAGATG | 3326 |
| 8690INT | TGAGGAATCACGTGTTTG | ATCGAATCTATCCATGAAGATG | 1746 |
| 90R3 | CATCATTGTTATCGCTTTCAC | GCAACCGCAGTTCCTGTT | 4912 |
| R395 | GCGGTACCCACTGATCTT | AGTCTACCAGGCATTCCGT | 6250 |
| 95100 | GAGGAGGGTCTGTCTAGATGT | TCGGAGATATCATAATCTGCGT | 4031 |
| 100DR | TTATAGTTGCTCCCGAAAGC | AAGAAGATGCGGTTGTCTTG | 2358 |
| ORF or Locus | Refseq bp | Refseq:SNP | Non-Synonymous SNP Amino Acid Change, Codons and (Reverse Complement Codons) | This Study or as Cited |
|---|---|---|---|---|
| DR-L DR6 | 6663 | G:GG | ||
| “ | 6665 | G:GG | Frameshift extension | [16] |
| DR-L repeat | 7623 | G:C | “ | “ |
| Repeats left end U | 8262 | AGGAG:A | ||
| “ | 8362 | C:CGCG | ||
| “ | 8518 | T:C | ||
| “ | 8559 | ACAACA:CAC | ||
| “ | 8591 | AT:A | ||
| “ | 8602 | GG:G | ||
| U10 | 18884 | C:Indel1 | Frameshift extension | [34,35] |
| U40 | 63871 | G:T | Thr:His (ACG:CAG) | |
| “ | 63872 | T:G | “ | |
| U42 | 70446 | A:ATC | HisCys:GlnLeuAsp (CACTGT:CAACTGCATG) | Jones & Teo 1993 in Genbank L20954 |
| “ | 70449 | G:GT | “ | Jones, M. in Genbank X92436 |
| U57 mcp | 92149 | C:G | Lys:Asn ( AAG:AAC) | |
| U58 tbp | 94068 | A:G | Met:Val ATG:GTG | |
| U83 chemokine | 123740 | C:G | AspGlu:GluGln GACGAG:GAGCAG | [15] |
| “ | 123741 | G:C | “ | “ |
| U86 IE2 | (128132) | (Indel G) | Frameshift U86/U87 | In RefSeq; [17] |
| R1 repeats | 128325 | C:T | ||
| “ | 128443 | C:T | ||
| “ | 128914 | G:A | ||
| “ | 129917 | A:ATCC | ||
| R2 repeats | 132312 | GG:G | ||
| R3 repeats | 137831 | C:G | ||
| “ | 137832 | G:C | ||
| “ | 137906 | C:G | ||
| “ | 137907 | G:C | ||
| U100 | 148154 | C:G | ||
| “ | 148155 | G:C | ||
| U100 gQ1 | 149553 | C:G | Arg:Ala (CGT: GCT) | |
| “ | 149554 | G:C | “ | |
| DR-R DR6 | 157898 | G:GG | Frameshift extension | [16] |
| “ | 157900 | G:GG | “ | |
| DR-R repeat | 158857 | G:C |
| ORF or Loci | RefSeq bp | RefSeq :SNP | Amino Acid Change, Codons and (Reverse Complement Codons) | % New Reference | % BAC Virus | % Passaged Virus | Comment, Citations, and Other Strains with SNPs |
|---|---|---|---|---|---|---|---|
| DR | 8016 | A:C | - | 87% | - | End T2 | |
| “ | 8077 | C:CGA | 96% | - | - | HHV-6A AJ KP257,584.1, GS KC465,951.1; CiHHV6A 2284 KT895,199.1, 1501 KT355,575.1; [37]; Sites cleavages after pac2; [26,31] | |
| “ | 8080 | G:GAC | - | 86% | - | CiHHV6A 2284 KT895,199.1, 5055, 5814 [16,24]; “ | |
| Uleft repeat | 8157 | G:GC | - | - | 78% | ||
| “ | 8558 | A:AC | 88% | AAC:A 99% | 90% | AC repeat | |
| “ | 8561 | AAC:A | - | 100% | 98% | HHV-6A AJ [15,31] | |
| “ | 8601 | CG:C | 97% | 87% | 86% | ||
| U2 | 8876 | G:T | Ala314:Glu(GCG:GAG) | - | - | 53% | |
| U19 UTR | 28,371 | C:CA | - | 52% | 72% | HHV-6A AJ | |
| U31 tegument | 45,667 | A:T | Asn173:Ile AAC:ATC | - | 73% | 96% | |
| U33 capsid | 52,148 | A:G | Tyr330:His(TAT:CAT) | - | 54% | 69% | |
| U39 gB* | 61,162 | T:C | Thr193:Ala(ACG:GCG) | - | 58% | 99% | |
| U41 dbp | 64,707 | C:T | Ala972:Thr (GCT:ACT) | - | - | 90% | HHV-6A GS KJ123,690.1 |
| ORI repeat | 67,674 | T:C | - | - | 54% | ||
| “ | 67,860 | T:C | - | - | 34% | HHV-6A GS KJ123,690.1 & KC465,951.1 | |
| U50 capsid | 81,583 | G:A | Ala258:Thr(GCA:ACA) | - | 59% | 78% | HHV-6A GS KC465,951.1 |
| “ | 82,158 | G:C | - | 67% | - | ||
| U73 obp | 109,214 | G:A | Ser297:Asn(AGT:AAT) | - | - | 99% | |
| U83 chemokine | 123,504 | G:GTT | f/s N-terminal extension | <1% | - | - | cDNA clone [13,14,15]; HHV-6A AJ KP257584; CiHHV6A: 5055 AIX09949, 23,090 AIX09956, 1501 KT355,575 |
| U86 IE2 | 126,126 | T:C | Asn395:Asp(AAT:GAT) | - | - | 99% | |
| “ | 127,092 | C:CTGA | Ser976:SerSer (TCA repeat ×9:×10) | - | 45% | - | 2 Ser insertion CiHHV6A 5055; 5814; [24] |
| R2 repeat | 131,092 | C:T | - | - | 56% | IE2 intron | |
| “ | 131,743 | G:C | - | - | 52% | “ | |
| “ | 131,745 | A:C | - | - | 62% | “ | |
| R2 region | 132,303 | TG:T | - | 64% | 56% | “ TGG:T HHV-6A AJ [31] | |
| R3 repeat | 140,872 | G:A | - | 96% | - | End 30× repeats from 137,775; latency IE1/2 intron [38] | |
| U95 | 142,969 | T:C | Trp10:Arg TGG:CGG | - | - | 54% | |
| “ | 142,979 | T:A | Ile13:Asn ATT:AAT | - | - | 53% | |
| “ | 143,591 | C:A | Ser217:Tyr TCC:TAC | - | - | 76% | |
| U100 gQ2 | 146,729 | C:T | C-terminal truncation; Trp186:stop (TGG:TGA) | <1% | - | - | [12]1 (in 7/8 gQ2 cDNA clones HHV-6A U1102) |
| “ | 146,862 | G:A | Ser42:Leu (TCG:TTG) | - | 60% | 99% | Not in HHV-6A HHV-6A U1102 from [39] |
| U100 gQ1 | 149,251 | Indel2:A | intron | - | 44% | 83% | HHV-6A AJ; CiHHV6A 2284, 5055, 5814 [24] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tweedy, J.G.; Escriva, E.; Topf, M.; Gompels, U.A. Analyses of Tissue Culture Adaptation of Human Herpesvirus-6A by Whole Genome Deep Sequencing Redefines the Reference Sequence and Identifies Virus Entry Complex Changes. Viruses 2018, 10, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/v10010016
Tweedy JG, Escriva E, Topf M, Gompels UA. Analyses of Tissue Culture Adaptation of Human Herpesvirus-6A by Whole Genome Deep Sequencing Redefines the Reference Sequence and Identifies Virus Entry Complex Changes. Viruses. 2018; 10(1):16. https://0-doi-org.brum.beds.ac.uk/10.3390/v10010016
Chicago/Turabian StyleTweedy, Joshua G., Eric Escriva, Maya Topf, and Ursula A. Gompels. 2018. "Analyses of Tissue Culture Adaptation of Human Herpesvirus-6A by Whole Genome Deep Sequencing Redefines the Reference Sequence and Identifies Virus Entry Complex Changes" Viruses 10, no. 1: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/v10010016