The Interaction Dynamics of Two Potato Leafroll Virus Movement Proteins Affects Their Localization to the Outer Membranes of Mitochondria and Plastids

 ,
,
Abstract
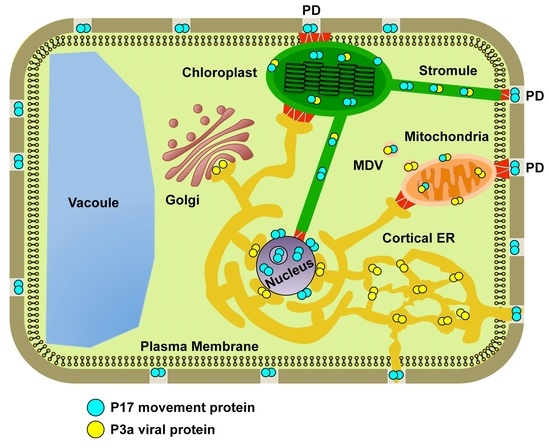
:
1. Introduction
2. Materials and Methods
2.1. Plant Growth Conditions and PLRV Infection
2.2. Affinity Purification (AP) of PLRV and Mass Spectrometry
2.3. MS Protein Identification and Label-Free Quantification
2.4. Bimolecular Fluorescent Complementation (BiFC) and Co-Localization Assays
2.5. Confocal Laser Scanning Microscopy
2.6. Immunodetection of PLRV and Fusion Proteins
3. Results
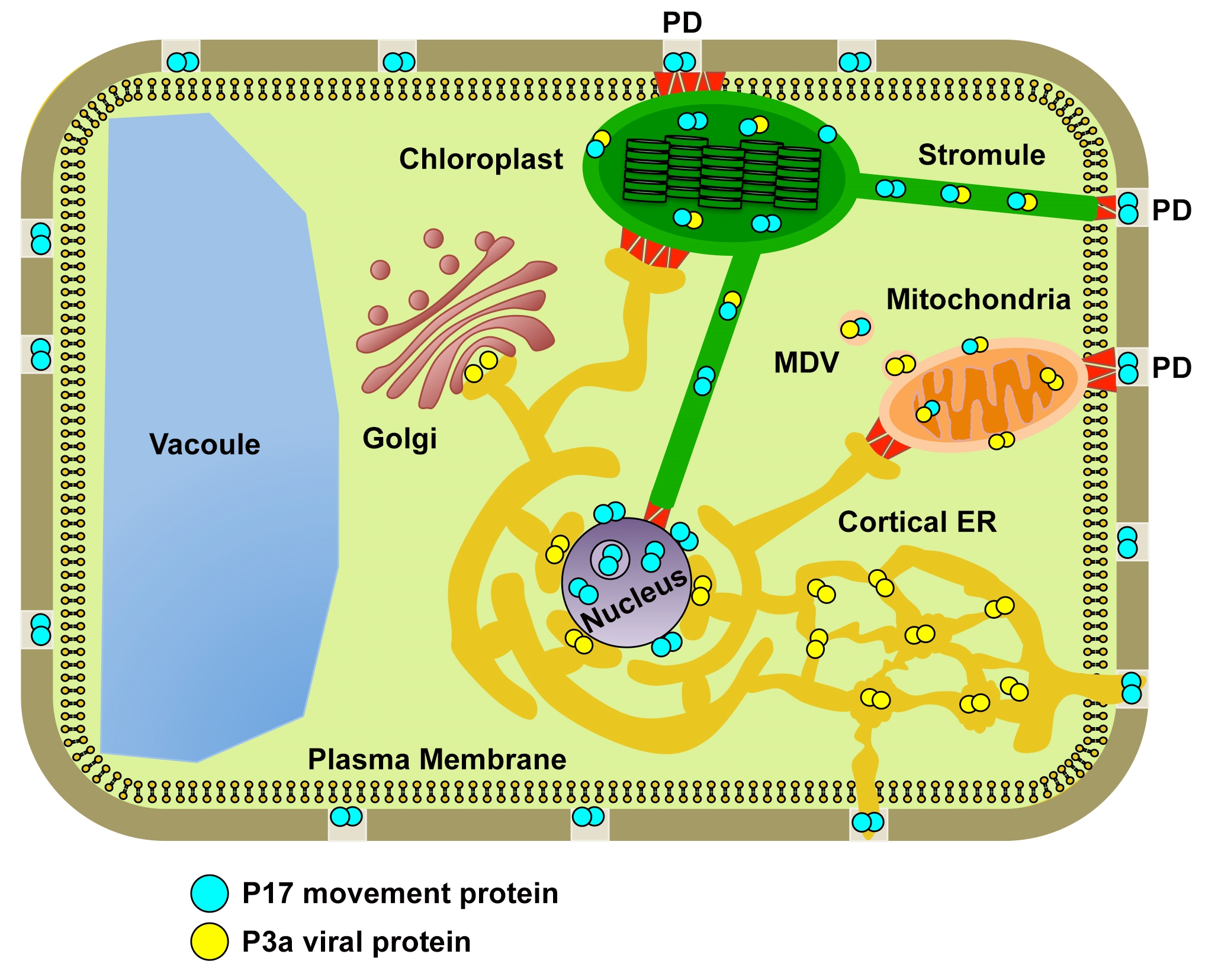
3.1. Expression Levels of the Coat Protein are Variable among Different PLRV Mutants
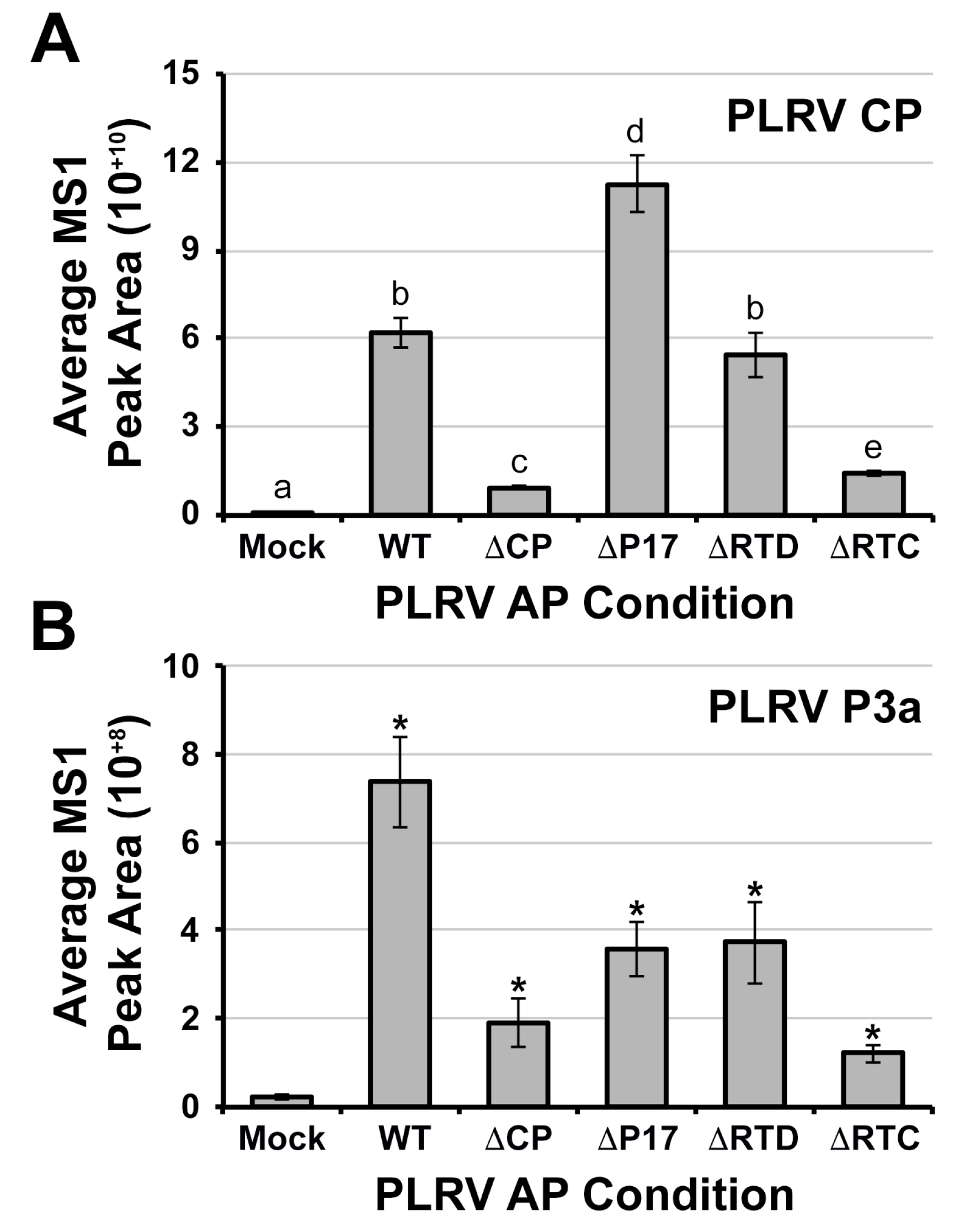
3.2. The Viral Protein P3a Complexes with PLRV and the Non-Incorporated Form of the RTP In Vivo
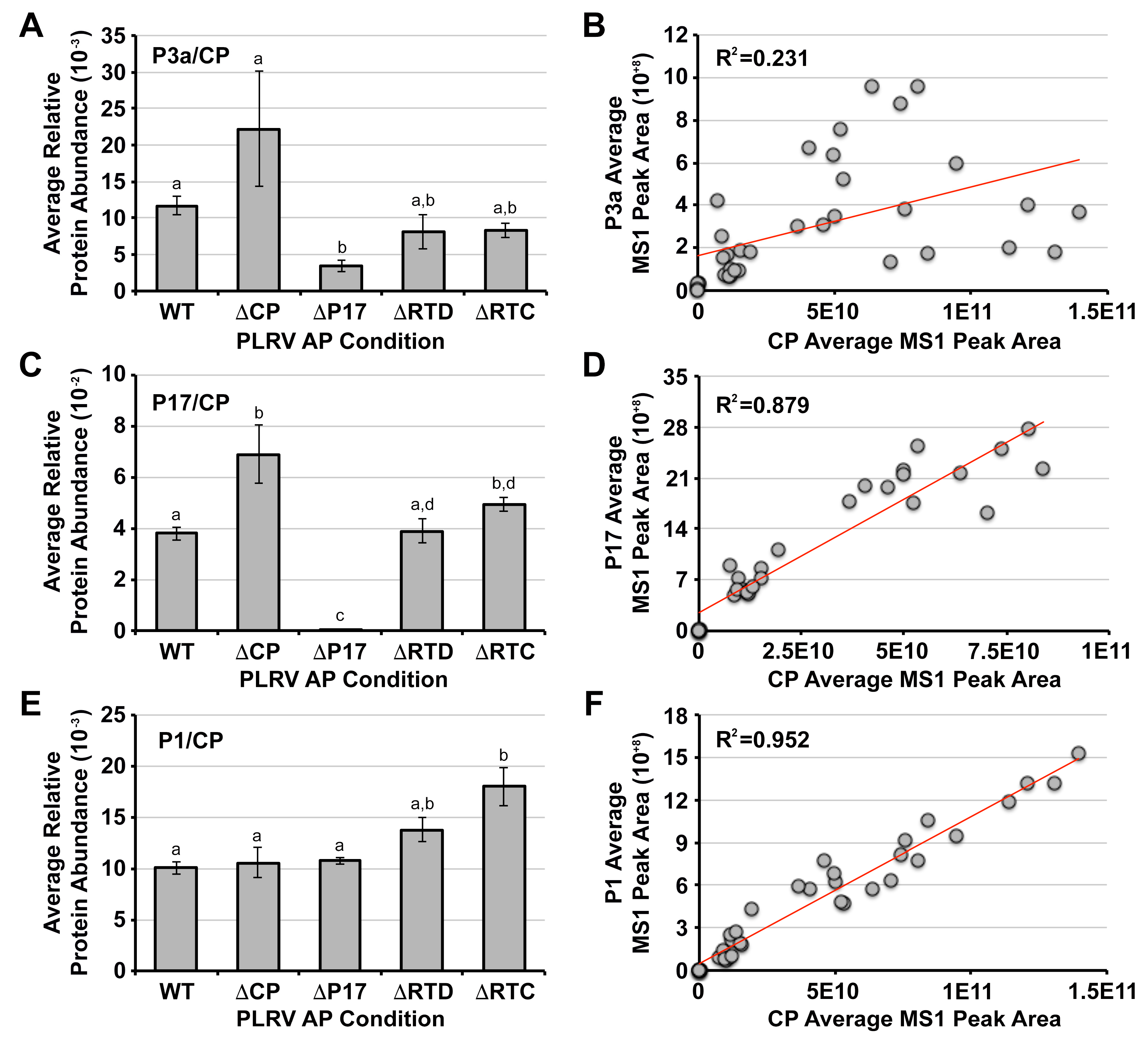
3.3. The Association of P3a with PLRV Is Partially Dependent on the Presence of the P17 Movement Protein
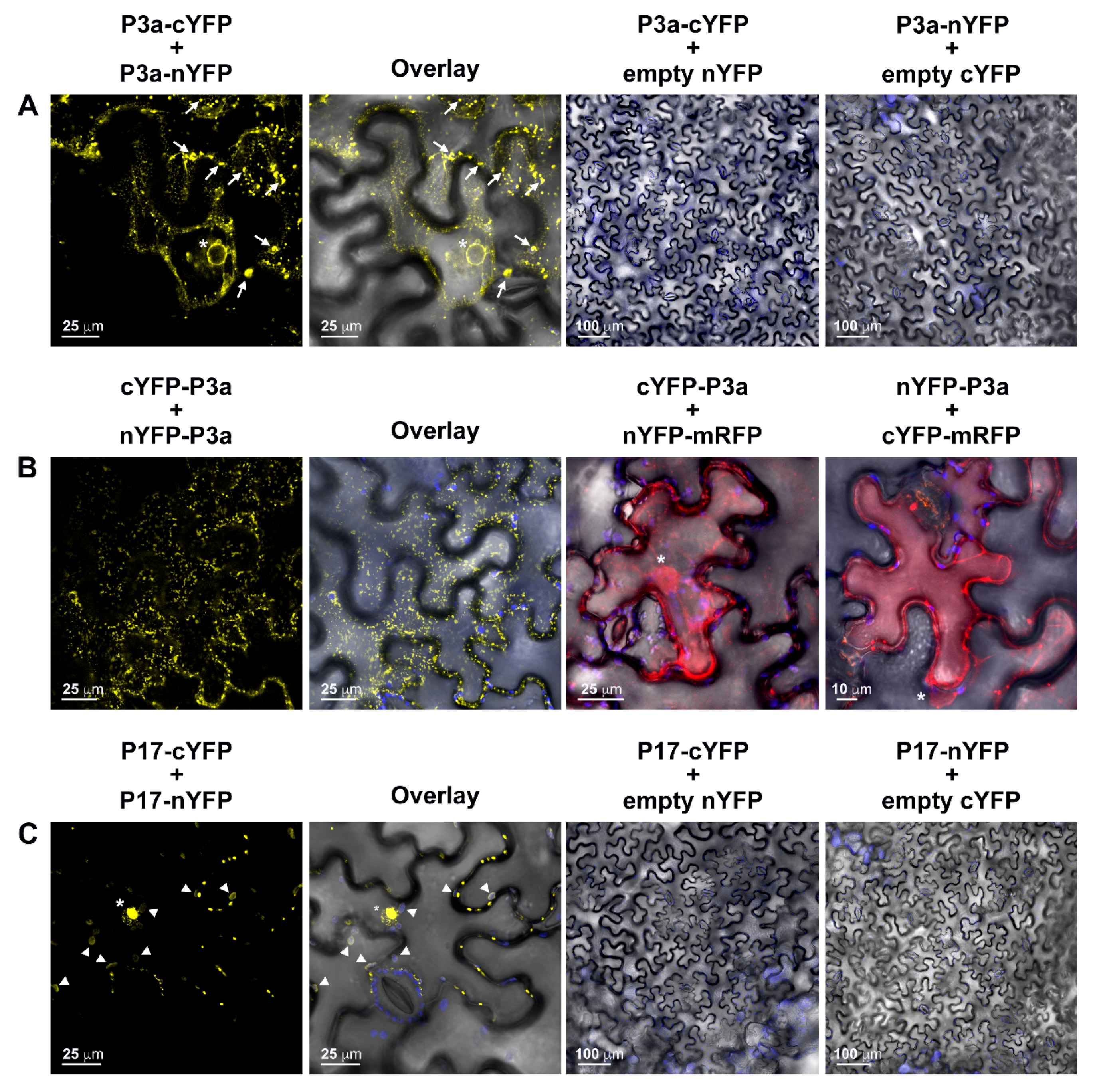
3.4. Characterization of PLRV P3a and P17 Homodimerization in Plant Cells
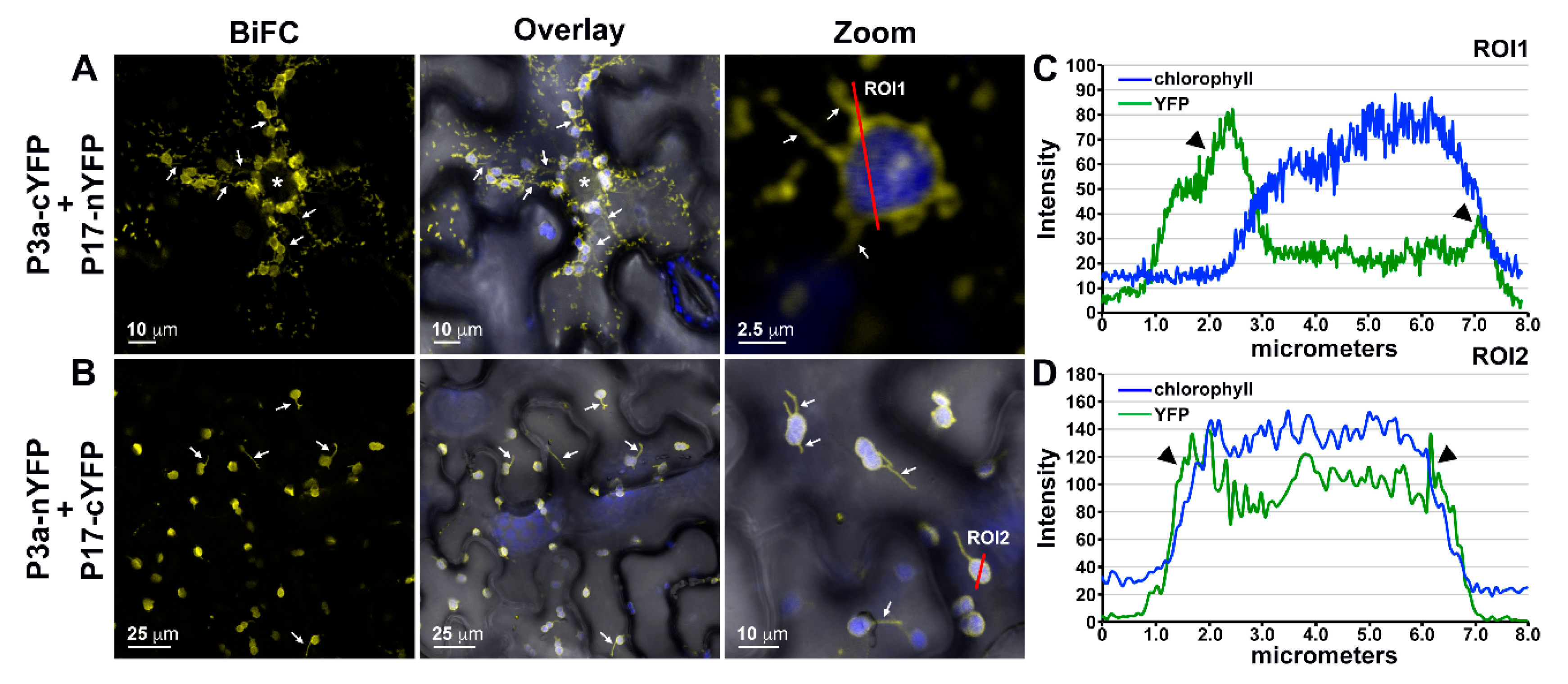
3.5. P3a Directly Interacts with P17 in Planta
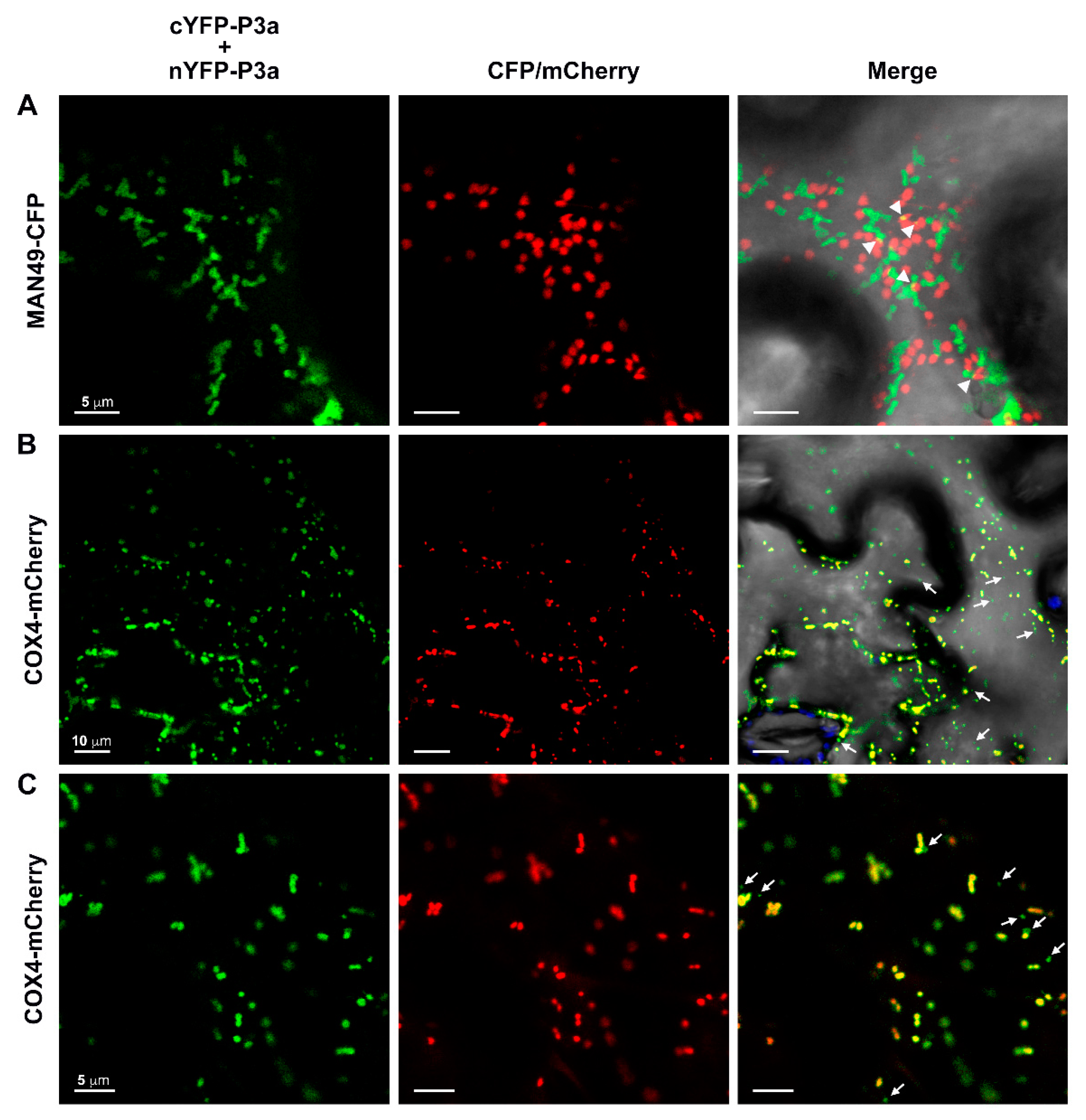
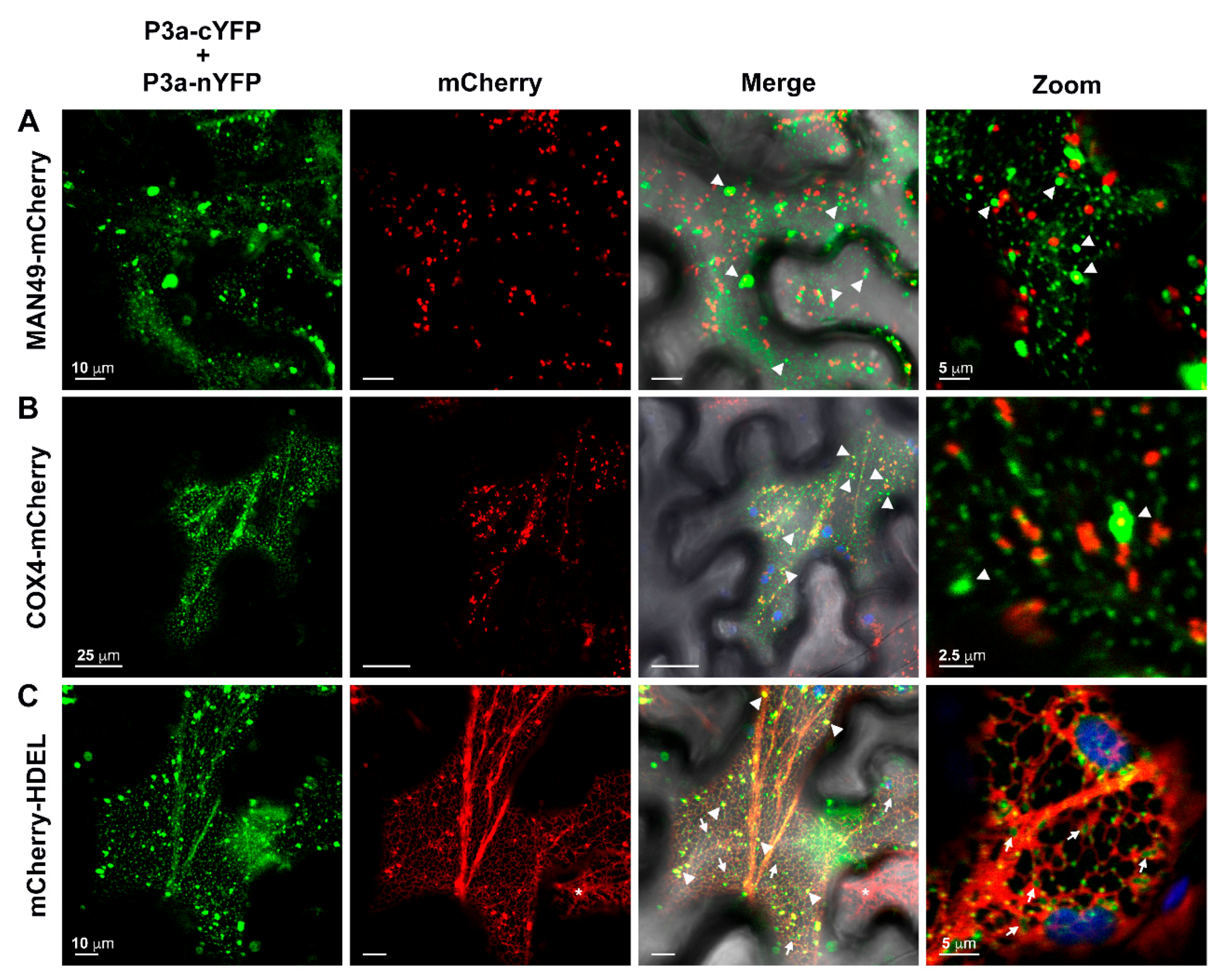
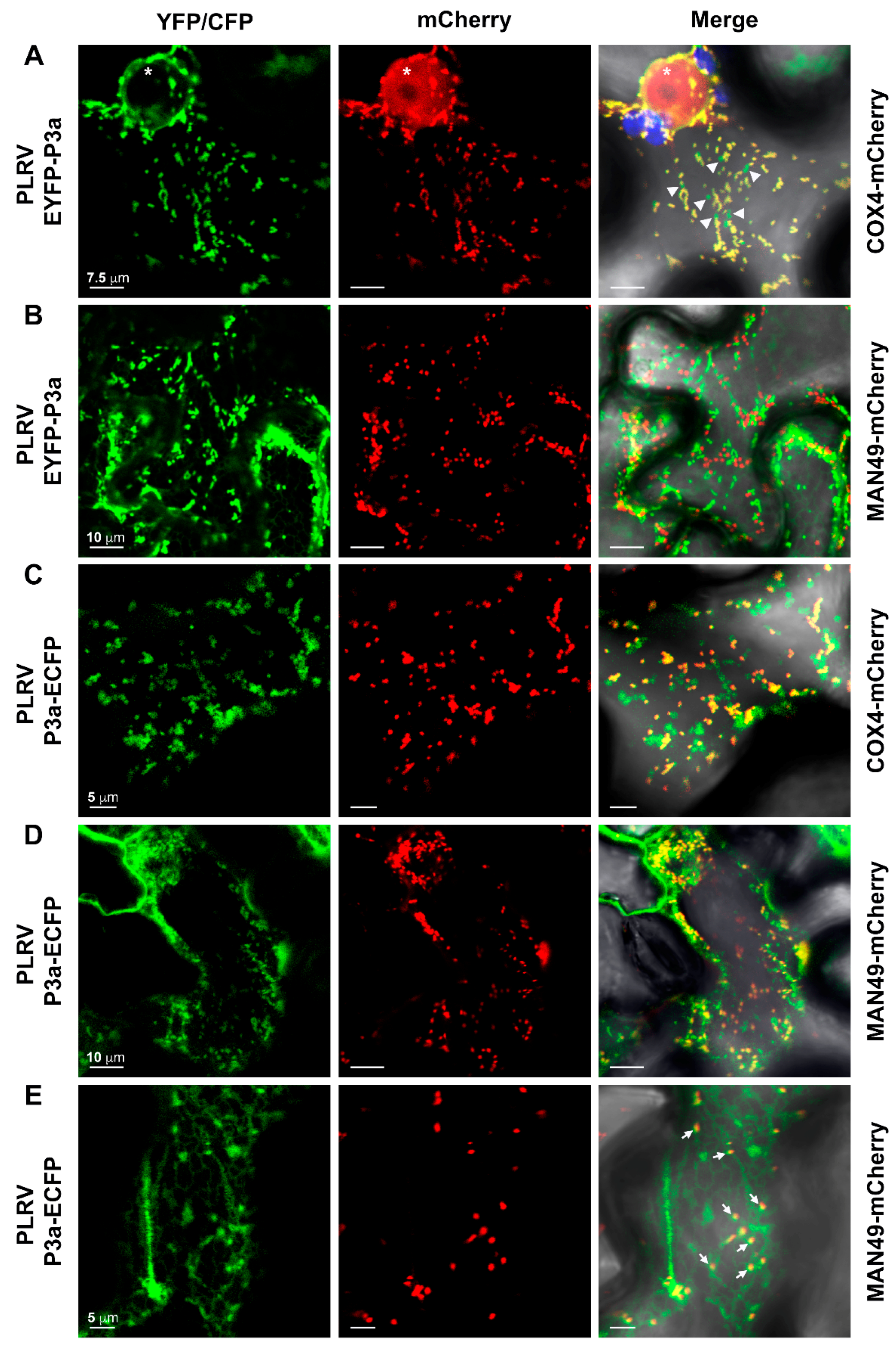
3.6. The P3a Homodimer and P3a-P17 Heterodimer Localize to the Outer Membrane of Mitochondria
3.7. Tag Orientation Influences the Subcellular Localization of PLRV P3a Fused to Full-Length Fluorescent Proteins
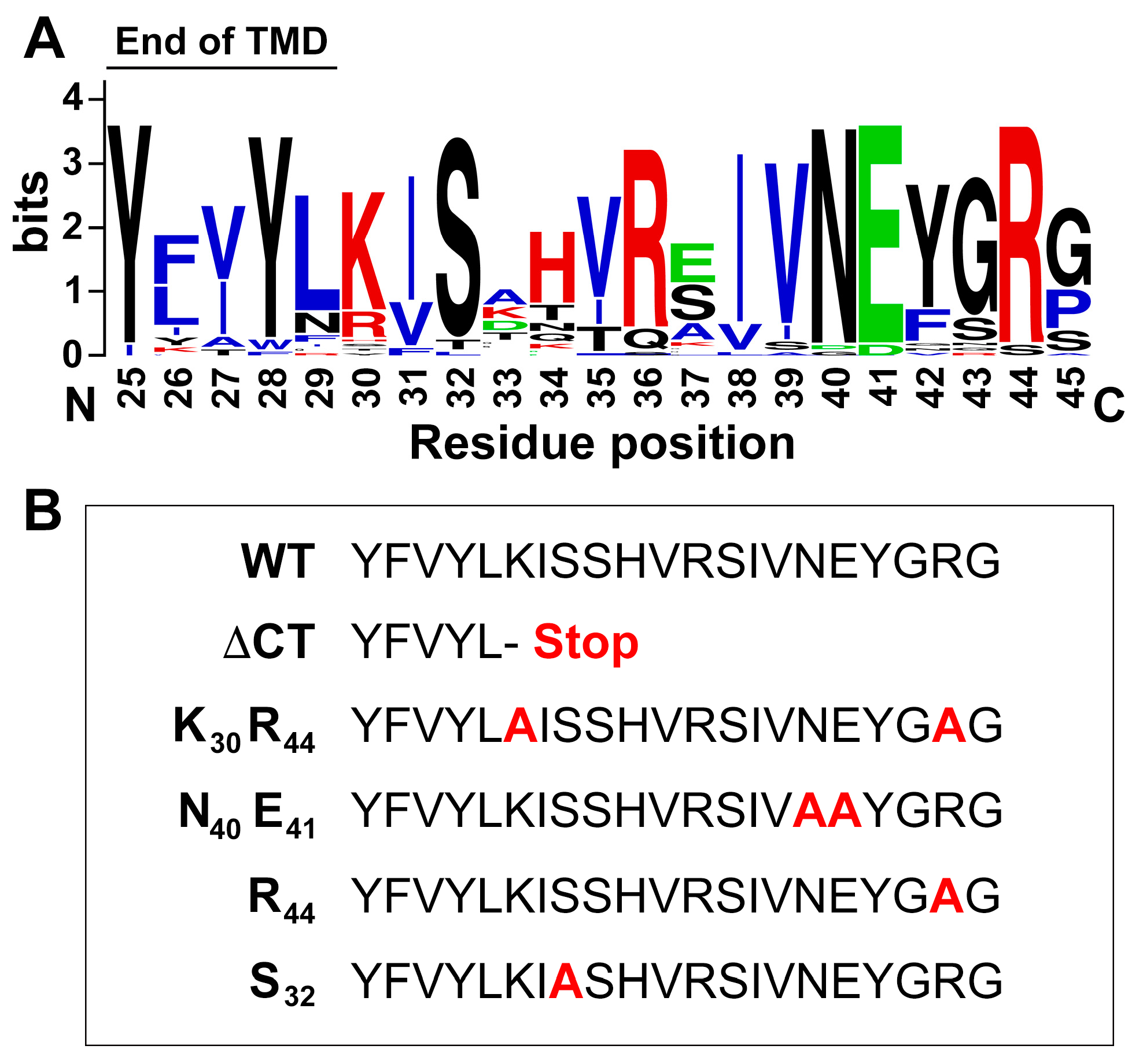
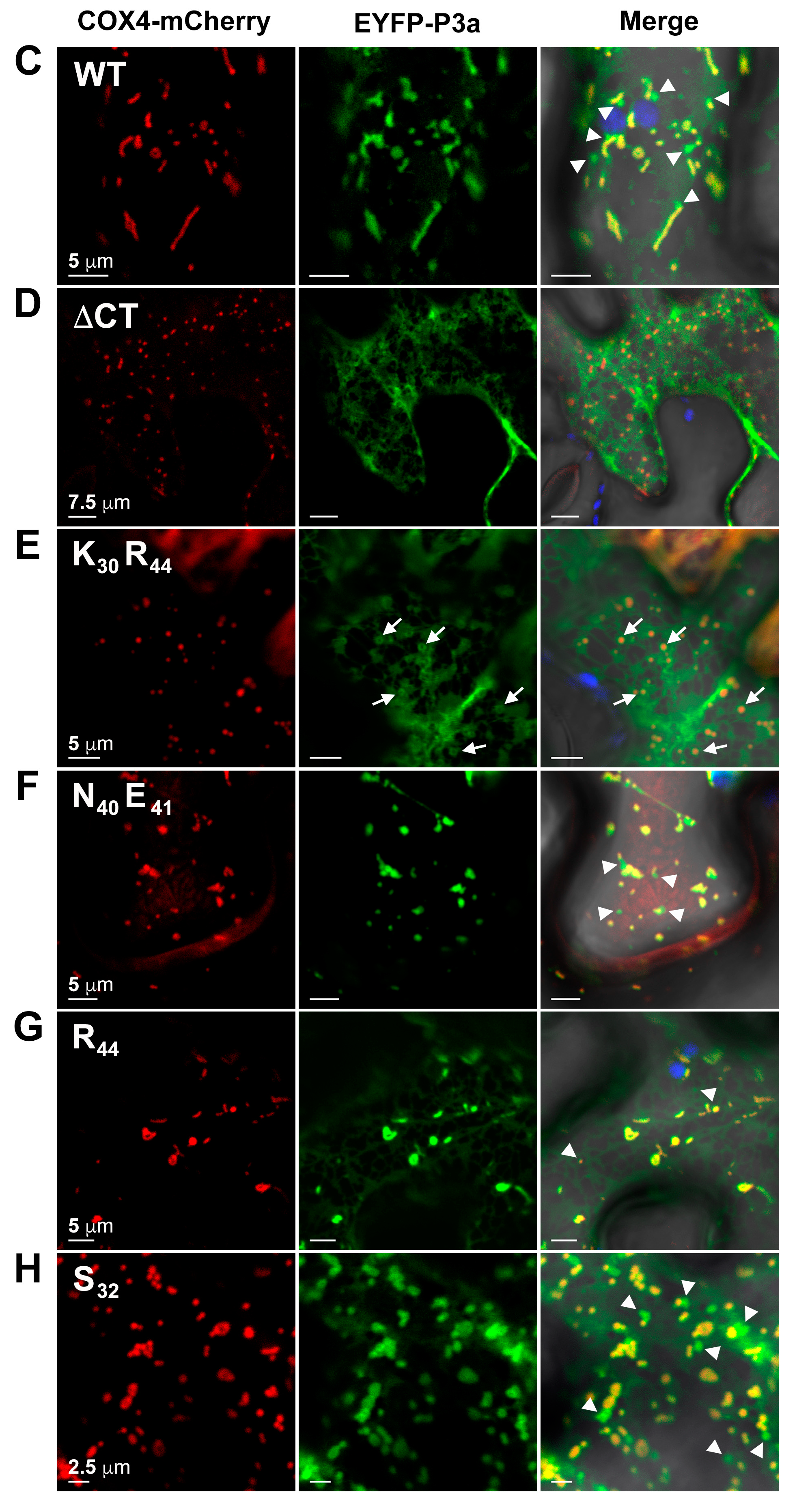
3.8. Basic Residues in the C-Terminus of PLRV P3a Facilitate Mitochondrial Localization
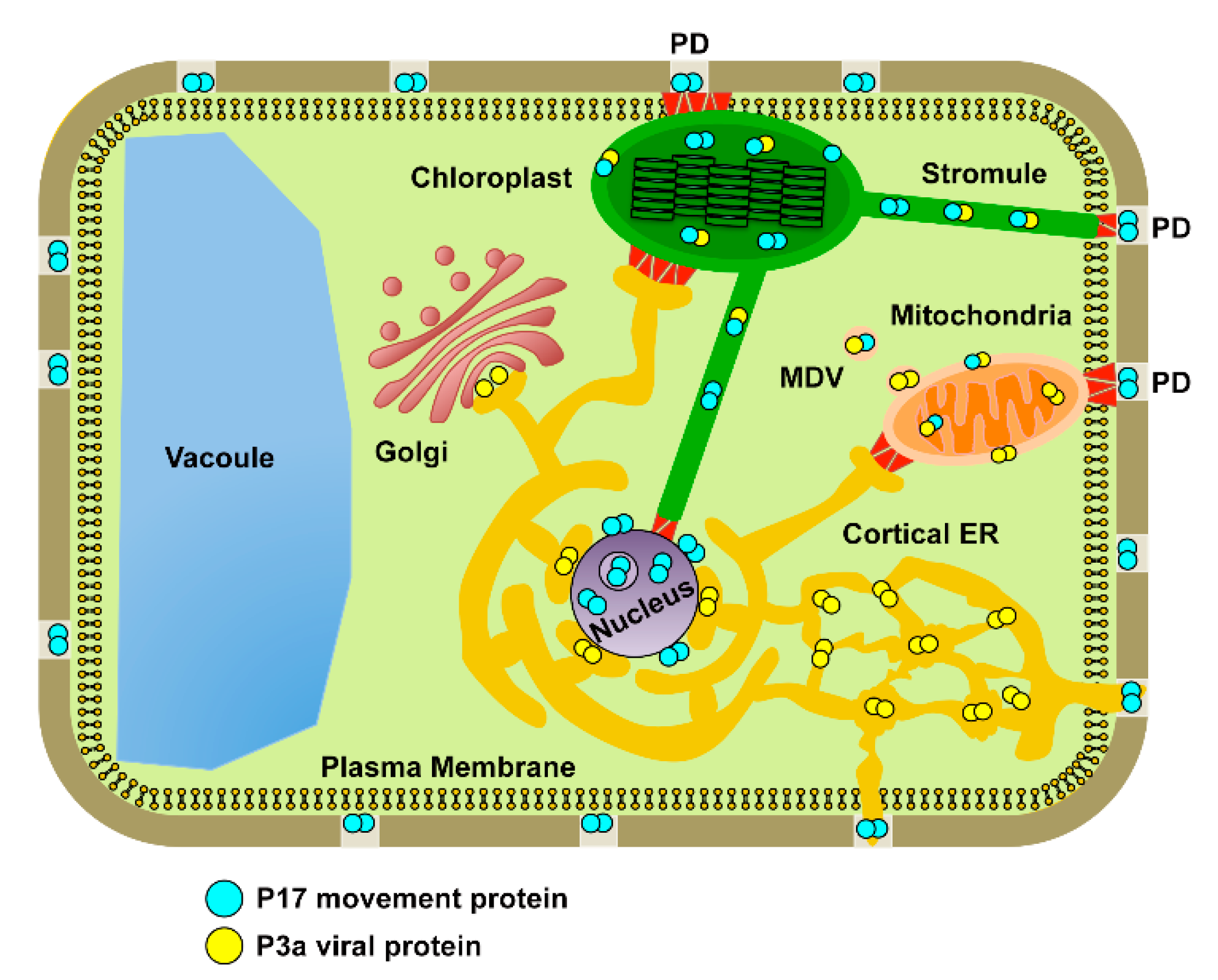
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeBlasio, S.L.; Chavez, J.D.; Alexander, M.; Ramsey, J.; Eng, J.K.; Mahoney, J.; Gray, S.M.; Bruce, J.E.; Cilia, M. Visualization of host-polerovirus interaction topologies using Protein Interaction Reporter Technology. J. Virol. 2015, 90, 1973–1987. [Google Scholar] [CrossRef] [PubMed]
- Benitez-Alfonso, Y.; Faulkner, C.; Ritzenthaler, C.; Maule, A.J. Plasmodesmata: Gateways to local and systemic virus infection. Mol. Plant Microbe Interact. 2010, 23, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Zheng, J.Y.; Lazarowitz, S.G. Synaptotagmin SYTA forms ER-Plasma membrane junctions that are recruited to plasmodesmata for plant virus movement. Curr. Biol. 2015, 25, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Nelson, R.S. The cell biology of Tobacco mosaic virus replication and movement. Front Plant Sci. 2013, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Verchot-Lubicz, J.; Torrance, L.; Solovyev, A.G.; Morozov, S.Y.; Jackson, A.O.; Gilmer, D. Varied movement strategies employed by Triple Gene Block–encoding viruses. Mol. Plant Microbe Interact. 2010, 23, 1231–1247. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Cilia, M.; Ghanim, M. Circulative, “Nonpropagative” Virus Transmission: An orchestra of virus, insect and plant derived instruments. Adv. Virus. Res. 2014, 89, 141–199. [Google Scholar] [PubMed]
- Gill, C.C.; Chong, J. Development of the infection in oat leaves inoculated with barley yellow dwarf virus. Virology 1975, 66, 440–453. [Google Scholar] [CrossRef]
- Brault, V.; Bergdoll, M.; Mutterer, J.; Prasad, V.; Pfeffer, S.; Erdinger, M.; Richards, K.E.; Ziegler-Graff, V. Effects of point mutations in the major capsid protein of beet western yellows virus on capsid formation, virus accumulation, and aphid transmission. J. Virol. 2003, 77, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Hipper, C.; Monsion, B.; Bortolamiol-Becet, D.; Ziegler-Graff, V.; Brault, V. Formation of virions is strictly required for turnip yellows virus long-distance movement in plants. J. Gen. Virol. 2014, 95, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, I.B.; Lee, L.; Ripoll, D.R.; Palukaitis, P.; Gildow, F.; Gray, S.M. Point mutations in the potato leafroll virus major capsid protein alter virion stability and aphid transmission. J. Gen. Virol. 2007, 88, 1821–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.; Kaplan, I.B.; Ripoll, D.R.; Liang, D.; Palukaitis, P.; Gray, S.M. A surface loop of the potato leafroll virus coat protein is involved in virion assembly, systemic movement, and aphid transmission. J. Virol. 2005, 79, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, S.; Erdinger, M.; Monsion, B.; Ziegler-Graff, V.; Brault, V. Both structural and non-structural forms of the readthrough protein of Cucurbit aphid-borne yellows virus are essential for effficient systemic infection of plants. PLoS ONE 2014, 9, e93448. [Google Scholar] [CrossRef] [PubMed]
- Brault, V.; Mutterer, J.; Scheidecker, D.; Simonis, M.T.; Herrbach, E.; Richards, K.; Ziegler-Graff, V. Effects of point mutations in the readthrough domain of the beet western yellows virus minor capsid protein on virus accumulation in planta and on transmission by aphids. J. Virol. 2000, 74, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Bruyere, A.; Brault, V.; Ziegler-Graff, V.; Simonis, M.T.; Van den Heuvel, J.F.; Richards, K.; Guilley, H.; Jonard, G.; Herrbach, E. Effects of mutations in the beet western yellows virus readthrough protein on its expression and packaging and on virus accumulation, symptoms, and aphid transmission. Virology 1997, 230, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Peter, K.A.; Gildow, F.; Palukaitis, P.; Gray, S.M. The C terminus of the polerovirus p5 readthrough domain limits virus infection to the phloem. J. Virol. 2009, 83, 5419–5429. [Google Scholar] [CrossRef] [PubMed]
- Peter, K.A.; Liang, D.; Palukaitis, P.; Gray, S.M. Small deletions in the potato leafroll virus readthrough protein affect particle morphology, aphid transmission, virus movement and accumulation. J. Gen. Virol. 2008, 89, 2037–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brault, V.; van den Heuvel, J.F.; Verbeek, M.; Ziegler-Graff, V.; Reutenauer, A.; Herrbach, E.; Garaud, J.C.; Guilley, H.; Richards, K.; Jonard, G. Aphid transmission of beet western yellows luteovirus requires the minor capsid read-through protein P74. EMBO J. 1995, 14, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Palukaitis, P.; Gray, S.M. Host-dependent requirement for the Potato leafroll virus 17-kda protein in virus movement. Mol. Plant Microbe Interact. 2002, 15, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Firth, A.E.; Miller, W.A.; Scheidecker, D.; Brault, V.; Reinbold, C.; Rakotondrafara, A.M.; Chung, B.Y.; Ziegler-Graff, V. Discovery of a small non-AUG-initiated ORF in poleroviruses and luteoviruses that is required for long-distance movement. PLoS Pathog. 2015, 11, e1004868. [Google Scholar] [CrossRef] [PubMed]
- Hofius, D.; Herbers, K.; Melzer, M.; Omid, A.; Tacke, E.; Wolf, S.; Sonnewald, U. Evidence for expression level-dependent modulation of carbohydrate status and viral resistance by the potato leafroll virus movement protein in transgenic tobacco plants. Plant J. 2001, 28, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, F.; Hofius, D.; Sonnewald, U. Intracellular trafficking of Potato leafroll virus movement protein in transgenic Arabidopsis. Traffic 2007, 8, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Link, K.; Vogel, F.; Sonnewald, U. PD Trafficking of potato leaf roll virus movement protein in Arabidopsis depends on site-specific protein phosphorylation. Front. Plant Sci. 2011, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, M.; Prüfer, D.; Tacke, E.; Rohde, W. The potato leafroll virus 17K movement protein is phosphorylated by a membrane-associated protein kinase from potato with biochemical features of protein kinase C. FEBS Lett. 1997, 400, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Tacke, E.; Schmitz, J.; Prufer, D.; Rohde, W. Mutational analysis of the nucleic acid-binding 17 kDa phosphoprotein of potato leafroll luteovirus identifies an amphipathic alpha-helix as the domain for protein-protein interactions. Virology 1993, 197, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Tacke, E.; Prufer, D.; Schmitz, J.; Rohde, W. The potato leafroll luteovirus 17K protein is a single-stranded nucleic acid-binding protein. J. Gen. Virol. 1991, 72, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhao, T.Y.; Li, Y.Y.; Xiang, H.Y.; Dong, S.W.; Zhang, Z.Y.; Wang, Y.; Li, D.W.; Yu, J.L.; Han, C.G. The conserved proline18 in the polerovirus P3a is important for brassica yellows virus systemic infection. Front. Microbiol. 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed]
- DeBlasio, S.L.; Johnson, R.; Mahoney, J.; Karasev, A.; Gray, S.M.; MacCoss, M.J.; Cilia, M. Insights into the polerovirus-plant interactome revealed by coimmunoprecipitation and mass spectrometry. Mol. Plant Microbe Interact. 2015, 28, 467–481. [Google Scholar] [CrossRef] [PubMed]
- DeBlasio, S.L.; Johnson, R.; Sweeney, M.M.; Karasev, A.; Gray, S.M.; MacCoss, M.J.; Cilia, M. Potato leafroll virus structural proteins manipulate overlapping, yet distinct protein interaction networks during infection. Proteomics 2015, 15, 2098–2112. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Gray, S.M.; Kaplan, I.; Palukaitis, P. Site-directed mutagenesis and generation of chimeric viruses by homologous recombination in yeast to facilitate analysis of plant-virus interactions. Mol. Plant Microbe Interact. 2004, 17, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ju, H.J.; DeBlasio, S.; Carino, E.J.; Johnson, R.; MacCoss, M.J.; Heck, M.; Miller, W.A.; Gray, S.M. A stem-loop structure in potato leafroll virus open reading frame 5 (ORF5) is essential for readthrough translation of the coat protein ORF stop codon 700 bases upstream. J. Virol. 2018, 92, e01544-17. [Google Scholar] [CrossRef] [PubMed]
- DeBlasio, S.L.; Johnson, R.S.; MacCoss, M.J.; Gray, S.M.; Cilia, M. Model system-guided protein interaction mapping for virus isolated from phloem tissue. J. Proteome Res. 2016, 15, 4601–4611. [Google Scholar] [CrossRef] [PubMed]
- DeBlasio, S.L.; Bereman, M.S.; Mahoney, J.; Thannhauser, T.W.; Gray, S.M.; MacCoss, M.J.; Cilia Heck, M. Evaluation of a bead-free coimmunoprecipitation technique for identification of virus-host protein interactions using high-resolution mass spectrometry. J. Biomol. Tech. 2017, 28, 111–121. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Falcone Ferreyra, M.L.; Casas, M.I.; Questa, J.I.; Herrera, A.L.; Deblasio, S.; Wang, J.; Jackson, D.; Grotewold, E.; Casati, P. Evolution and expression of tandem duplicated maize flavonol synthase genes. Front. Plant Sci. 2012, 3, 101. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Bui, H.T.; Pautler, M.; Llaca, V.; Johnston, R.; Lee, B.H.; Kolbe, A.; Sakai, H.; Jackson, D. A maize glutaredoxin gene, Abphyl2, regulates shoot meristem size and phyllotaxy. Plant Cell 2015, 27, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Wu, J.; Lu, L.; Xu, Y.; Zhou, X. Interaction between Rice stripe virus disease-specific protein and host PsbP enhances virus symptoms. Mol. Plant 2014, 7, 691–708. [Google Scholar] [CrossRef] [PubMed]
- Earley, K.W.; Haag, J.R.; Pontes, O.; Opper, K.; Juehne, T.; Song, K.; Pikaard, C.S. Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 2006, 45, 616–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castañeda, A.; Reddy, J.D.; El-Yacoubi, B.; Gabriel, D.W. Mutagenesis of all eight avr genes in Xanthomonas campestris pv. campestris had no detected effect on pathogenicity, but one avr gene affected race specificity. Mol. Plant Microbe Interact. 2005, 18, 1306–1317. [Google Scholar]
- DeBlasio, S.L.; Rebelo, A.R.; Parks, K.; Gray, S.; Cilia, M. Disruption of chloroplast function through downregulation of phytoene desaturase enhances the systemic accumulation of an aphid-borne, phloem-restricted virus. Mol. Plant Microbe Interact. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.K.; Cai, X.; Nebenfuhr, A. A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 2007, 51, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Harrison, M.J. A set of fluorescent protein-based markers expressed from constitutive and arbuscular mycorrhiza-inducible promoters to label organelles, membranes and cytoskeletal elements in Medicago truncatula. Plant J. 2014, 80, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Larsen, B.; Lin, Z.Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.C.; Nesvizhskii, A.I. SAINT: Probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Griffing, L.R.; Lin, C.; Perico, C.; White, R.R.; Sparkes, I. Plant ER geometry and dynamics: Biophysical and cytoskeletal control during growth and biotic response. Protoplasma 2017, 254, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, J.; Kim, K.; Lee, K.J.; Lee, W.H.; Ju, H.J. Localization of Barley yellow dwarf virus Movement Protein Modulating Programmed Cell Death in Nicotiana benthamiana. Plant Pathol. J. 2017, 33, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Xia, Z.; Zhang, Y.; Wen, Y.; Wang, D.; Brandenburg, K.; Harris, F.; Pheonix, D.A. Interaction between the movement protein of barley yellow dwarf virus and the cell nuclear envelope: Role of a putative amphiphilic α-helix at the N-terminus of the movement protein. Biopolymers 2005, 79, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.R.; Hines, K.M. Stromules: Probing Formation and Function. Plant Physiol. 2018, 176, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Stussi-Garaud, C.; Tacke, E.; Prufer, D.; Rohde, W.; Rohfritsch, O. In situ localization of the putative movement protein (PR17) from potato leafroll luteovirus (PLRV) in infected and transgenic potato plants. Virology 1997, 235, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Saint-Jore-Dupas, C.; Nebenfuhr, A.; Boulaflous, A.; Follet-Gueye, M.L.; Plasson, C.; Hawes, C.; Driouich, A.; Faye, L.; Gomord, V. Plant N-glycan processing enzymes employ different targeting mechanisms for their spatial arrangement along the secretory pathway. Plant Cell 2006, 18, 3182–3200. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Fujimoto, M.; Katayama, K.; Yamaoka, S.; Tsutsumi, N.; Arimura, S. Formation of mitochondrial outer membrane derived protrusions and vesicles in Arabidopsis thaliana. PLoS ONE 2016, 11, e0146717. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.G.; Brandizzi, F.; Hawes, C.; Nakano, A. Vesicles versus tubes: Is endoplasmic reticulum-Golgi transport in plants fundamentally different from other eukaryotes? Plant Physiol. 2015, 168, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Lang-Pauluzzi, I. The behaviour of the plasma membrane during plasmolysis: A study by UV microscopy. J. Microsc. 2000, 198, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Oparka, K.J.; Prior, D.A.M.; Crawford, J.W. Behaviour of plasma membrane, cortical ER and plasmodesmata during plasmolysis of onion epidermal cells. Plant Cell Environ. 1994, 17, 163–171. [Google Scholar] [CrossRef]
- Lerch-Bader, M.; Lundin, C.; Kim, H.; Nilsson, I.; von Heijne, G. Contribution of positively charged flanking residues to the insertion of transmembrane helices into the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 4127–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Laliberté, J.-F. Membrane association for plant virus replication and movement. In Current Research Topics in Plant Virology; Wang, A., Zhou, X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 67–85. [Google Scholar]
- Verchot, J. Wrapping membranes around plant virus infection. Curr. Opin. Virol. 2011, 1, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Cowan, G.H.; Ziegler, A.; Roberts, A.G.; Oparka, K.J.; Torrance, L. Two plant-viral movement proteins traffic in the endocytic recycling pathway. Plant Cell 2005, 17, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.J.; Ye, C.M.; Verchot-Lubicz, J. Mutational analysis of PVX TGBp3 links subcellular accumulation and protein turnover. Virology 2008, 375, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.R.; Niewiadomski, S.; Prosser, S.W.; Krell, P.; Meng, B. Subcellular localization of the triple gene block proteins encoded by a foveavirus infecting grapevines. Virus Res. 2008, 138, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, A.G.; Stroganova, T.A.; Zamyatnin, A.A., Jr.; Fedorkin, O.N.; Schiemann, J.; Morozov, S.Y. Subcellular sorting of small membrane-associated triple gene block proteins: TGBp3-assisted targeting of TGBp2. Virology 2000, 269, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Lee, S.C.; Wang, C.W. Viral protein targeting to the cortical endoplasmic reticulum is required for cell-cell spreading in plants. J. Cell Biol. 2011, 193, 521–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepardson, S.; Esau, K.; McCrum, R. Ultrastructure of potato leaf phloem infected with potato leafroll virus. Virology 1980, 105, 379–392. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W. Cationic peptides: Effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 2001, 1, 156–164. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2, 1018.1–1018.5. [Google Scholar] [CrossRef]
- Natesan, S.K.; Sullivan, J.A.; Gray, J.C. Stromules: A characteristic cell-specific feature of plastid morphology. J. Exp. Bot. 2005, 56, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.; Chakraborty, S. Chloroplast: The Trojan horse in plant-virus interaction. Mol. Plant Pathol. 2017, 19, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Caplan, J.L.; Kumar, A.S.; Park, E.; Padmanabhan, M.S.; Hoban, K.; Modla, S.; Czymmek, K.; Dinesh-Kumar, S.P. Chloroplast stromules function during innate immunity. Dev. Cell 2015, 34, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Krenz, B.; Windeisen, V.; Wege, C.; Jeske, H.; Kleinow, T. A plastid-targeted heat shock cognate 70kDa protein interacts with the Abutilon mosaic virus movement protein. Virology 2010, 401, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Krenz, B.; Jeske, H.; Kleinow, T. The induction of stromule formation by a plant DNA-virus in epidermal leaf tissues suggests a novel intra- and intercellular macromolecular trafficking route. Front. Plant Sci. 2012, 3, 291. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, K.; Kasahara, M.; Kiyosue, T.; Kagawa, T.; Suetsugu, N.; Takahashi, F.; Kanegae, T.; Niwa, Y.; Kadota, A.; Wada, M. CHLOROPLAST UNUSUAL POSITIONING1 is essential for proper chloroplast positioning. Plant Cell 2003, 15, 2805–2815. [Google Scholar] [CrossRef] [PubMed]
- Eigenbrode, S.D.; Ding, H.; Shiel, P.; Berger, P.H. Volatiles from potato plants infected with potato leafroll virus attract and arrest the virus vector, Myzus persicae (Homoptera: Aphididae). Proc. Biol. Sci. 2002, 269, 455–460. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N. benthamiana AP c | S. tuberosum AP d | ||||
|---|---|---|---|---|---|
| Organelle a | Host Protein b | Fold Enrichment e | SAINT f | Fold Enrichment e | SAINT f |
| Plastid | TOC75-III | 200.6 | 1 | +/− | 1 |
| TOC34 | 35.3 | 1 | +/− | 0.92 | |
| TOC159 | 33.4 | 1 | nd | nd | |
| TOC132 | +/− | 0.8 | nd | nd | |
| OEP16 | +/− | 0.84 | +/− | 0.64 | |
| OEP37 | +/− | 0.82 | nd | nd | |
| CHUP1 | +/− | 0.85 | nd | nd | |
| HSP12 | +/− | 0.98 | nd | nd | |
| Mitochondria | VDAC1 | +/− | 0.98 | 1.9 | 0.61 |
| DRP1E | 10.2 | 1 | 7.7 | 1 | |
| Nucleus | NOP56 | 55.7 | 0.96 | +/− | 0.64 |
| NOP5-2 | 37.8 | 1 | nd | nd | |
| AT-IMP | 53.9 | 0.93 | +/− | 0.23 | |
| KPNB1 | +/− | 0.94 | +/− | 0.44 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeBlasio, S.L.; Xu, Y.; Johnson, R.S.; Rebelo, A.R.; MacCoss, M.J.; Gray, S.M.; Heck, M. The Interaction Dynamics of Two Potato Leafroll Virus Movement Proteins Affects Their Localization to the Outer Membranes of Mitochondria and Plastids. Viruses 2018, 10, 585. https://0-doi-org.brum.beds.ac.uk/10.3390/v10110585
DeBlasio SL, Xu Y, Johnson RS, Rebelo AR, MacCoss MJ, Gray SM, Heck M. The Interaction Dynamics of Two Potato Leafroll Virus Movement Proteins Affects Their Localization to the Outer Membranes of Mitochondria and Plastids. Viruses. 2018; 10(11):585. https://0-doi-org.brum.beds.ac.uk/10.3390/v10110585
Chicago/Turabian StyleDeBlasio, Stacy L., Yi Xu, Richard S. Johnson, Ana Rita Rebelo, Michael J. MacCoss, Stewart M. Gray, and Michelle Heck. 2018. "The Interaction Dynamics of Two Potato Leafroll Virus Movement Proteins Affects Their Localization to the Outer Membranes of Mitochondria and Plastids" Viruses 10, no. 11: 585. https://0-doi-org.brum.beds.ac.uk/10.3390/v10110585