Who’s Driving? Human Cytomegalovirus, Interferon, and NFκB Signaling

Department of Biochemistry and Biophysics, University of Rochester Medical Center, Rochester, NY 14642, USA
*
Author to whom correspondence should be addressed.
Viruses 2018, 10(9), 447; https://0-doi-org.brum.beds.ac.uk/10.3390/v10090447
Submission received: 3 August 2018
/
Revised: 17 August 2018
/
Accepted: 18 August 2018
/
Published: 21 August 2018
(This article belongs to the Special Issue Recent Advances in Cytomegalovirus Research)
{kind=link}
{kind=link}
Abstract
:As essential components of the host’s innate immune response, NFκB and interferon signaling are critical determinants of the outcome of infection. Over the past 25 years, numerous Human Cytomegalovirus (HCMV) genes have been identified that antagonize or modulate the signaling of these pathways. Here we review the biology of the HCMV factors that alter NFκB and interferon signaling, including what is currently known about how these viral genes contribute to infection and persistence, as well as the major outstanding questions that remain.
1. Introduction
Human cytomegalovirus (HCMV) is a widespread opportunistic pathogen that causes disease in a variety of immunosuppressed populations, including the elderly, cancer patients, and AIDS patients [1,2]. HCMV infection also causes significant morbidity in transplant recipients, and it is a major cause of kidney, liver, heart and bone marrow transplant rejection [3]. HCMV infection is also a significant cause of congenital disability, as roughly 5 out of every 1000 infants born each year are infected with HCMV, and approximately 10% of that population will experience neurological symptoms [2,4,5].
The relationship between HCMV and host immunity, including recognition, priming, and the subsequent host response, is a major determinant of HCMV pathogenesis. The earliest events of this response typically involve innate immune sensing of infection in non-immune cells, induction of an anti-viral state, and secretion of anti-viral paracrine factors that both help neighboring cells resist infection as well as recruit and activate professional immune cells. Interferon and NFκB signaling are two signal transduction cascades integral to these processes that are frequently targeted by viral infection to ensure persistence. The past 25 years have seen great progress in identifying numerous HCMV viral factors that modulate these innate immune pathways. However, many questions remain about both the specific biochemical mechanisms involved as well as the contexts through which they contribute to viral pathogenesis, replication, and persistence. Given their importance to infectious outcomes, further elucidating the diverse roles of these viral innate immune modulators remains a high priority to further our understanding of viral biology, while also potentially providing fertile ground for therapeutic development.
2. HCMV and Interferon Signaling
Interferons (IFN) are a broad class of cytokines initially recognized for their ability to protect cells from viral infection. Anti-viral IFN signaling in response to infection is capable of inducing a variety of host cell defenses targeting various aspects of virus biology, including global inhibition of translation via activation of protein kinase R (PKR), induction of RNAse L-mediated degradation of viral RNA, production of anti-viral nitric oxide through inducible nitric oxide synthase (iNOS), depletion of tryptophan via the indoleamine 2,3 dioxygenase (IDO) pathway, and upregulation of antigen presentation via major histocompatibility (MHC) complex constituents [6,7,8,9,10]. IFN signaling is sub-classified into Type I, Type II, and Type III groups based on their specific signaling architectures and outcomes. The majority of interferon-induced anti-viral activity is dependent on the diverse activities of Type I IFNs, which in humans include IFNκ, IFNε, IFNω, IFNβ, and a host of IFNα isoforms derived from 13 separate genes [11]. The activities of Type I IFNs induce the expression of a large set of IFN-stimulated genes (ISGs) that are critical for rendering the host cell environment less permissive for viral infection. In addition to the direct effects of Type I cytokines on ISG induction, these IFNs have been implicated in coordinated innate immune effector processes including Natural Killer (NK) cell activation, dendritic cell (DC) maturation, and T-cell differentiation [12]. In contrast to the diverse signaling of Type I IFNs, Type II IFN signaling is comprised entirely of the activities of a single cytokine: IFNγ. While IFNγ is also capable of activating ISG expression, its primary function appears to center around enabling the activation, recruitment, and survival of a diverse array of immune cells [13]. The final interferon subtype, Type III, was discovered more recently. Like Type I IFN signaling, Type III IFN signaling plays an important role in host cell defense against viral infection, but it relies on a different subset of IFNs (IFN-λ1, IFN-λ2, and IFN-λ3) and a distinct, heterodimeric IL28Rα/IL10Rβ receptor to induce the expression of anti-viral ISGs. As expression of the Type III IFN receptor is limited to epithelial cell populations, Type III IFN signaling is restricted to a more niche biological context than Type I signaling [14]. HCMV gene products that interact directly with Type III signaling have yet to be described. Canonical signaling of all three IFN subtypes occurs through the activation of the JAK-STAT pathway, but each subtype coordinates with unique cellular receptors and recruits specific STAT proteins that tailor downstream responses to respective Type I, II, and III IFN signaling inputs (reviewed in [15]).
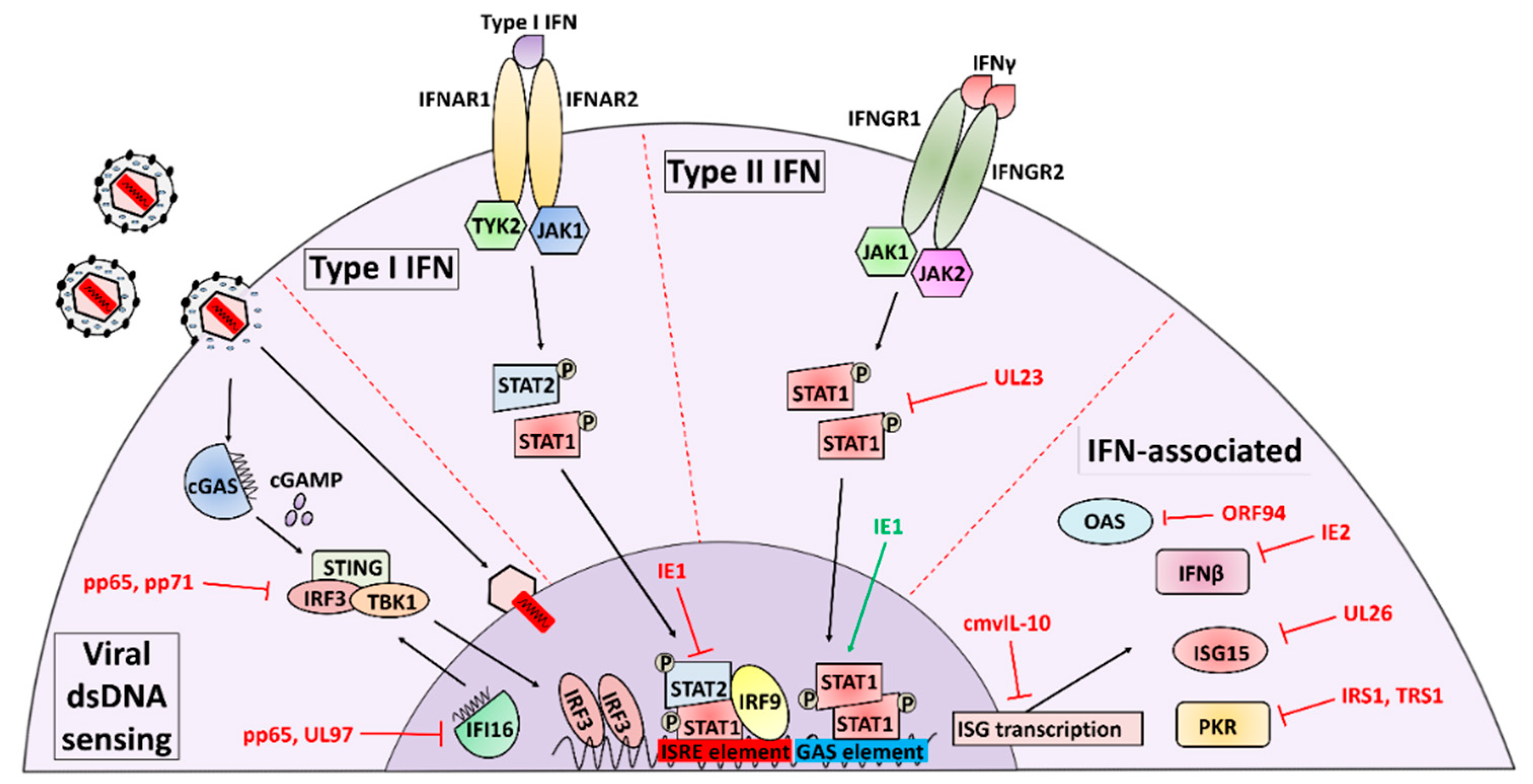
The IFN response to HCMV infection is complex, with multiple distinct mechanisms of IFN activation and temporal peaks of IFN activity occurring over the viral life cycle. The initial interferon response to HCMV infection is triggered when the cell detects viral attachment and entry, resulting in an early induction of IFN synthesis and secretion [16]. The list of cellular sensors that detect and are activated by HCMV binding and entry continues to expand, and includes the TLR2 (toll-like receptor) and CD14 receptors that interact with viral gB and gH [17,18] as well as the intracellular dsDNA sensors Z-DNA binding protein 1 (ZBP1) [19], TLR9 [20], and cGAS [20], all of which detect the presence of the viral genome in the host cell. Seemingly in response to these cellular defenses, HCMV has evolutionarily developed a suite of IFN countermeasures that occupy a significant portion of viral coding potential (summarized in Figure 1 and reviewed in [12,21,22]).
2.1. The HCMV Tegument Proteins and Interferon Modulation
Given that the IFN response to infection is a swift anti-viral response set in motion by the first interactions between virion glycoproteins and cellular receptors, it is logical that HCMV encodes viral effectors that immediately antagonize these early anti-viral events, and downregulation of IFN signaling during HCMV infection is well-documented [23,24,25,26]. Many of these early viral anti-interferon factors are tegument proteins that are delivered to the cellular cytoplasm upon initial envelope fusion. One of the first of these HCMV-encoded IFN modulators described was the UL83-encoded tegument protein pp65, which is packaged into the virion in such abundance that it is the major constituent of the infectious particle [27]. The pp65 protein plays a major role in inhibiting both innate and adaptive immune responses to HCMV infection by interfering with antigen presentation [28] and NK cell activation [29]. HCMV pp65 was initially characterized as a Type I IFN inhibitor through early reports utilizing a UL83-deletion virus that induced IFN-associated transcriptional patterns during infection. Varying hypotheses of pp65’s mechanistic function emerged as some groups reported that pp65 was sufficient to block activation and nuclear localization of IRF3, a primary mediator of Type I IFN signaling [30], while others reported no change in IRF3 activity but observed that pp65 loss significantly increased IRF1 and NFκB nuclear localization [31]. Subsequent studies suggested that the pp65 deletion mutant virus exhibited impaired expression of other important HCMV genes, including the immediate-early protein 2 (IE2), and found that a less-disruptive mutation of the pp65 ORF maintained viral inhibition of Type I IFN signaling during infection [32], indicating that pp65 may be more dispensable for inhibition of IFN signaling than was initially presumed.
The question of how pp65 contributes to HCMV-mediated inhibition of IFN activity is largely still unresolved. While some IFN phenotypes originally attributed to pp65 appear to be due to the actions of other viral gene products such as IE2, new roles for pp65 are continually being revealed, such as the interaction between pp65 and activators of the stimulator of interferon genes (STING), an IFN inducer. Signaling via STING and the dsDNA sensor cGAS has emerged as an important cellular defense against viral infection [33]. A novel binding interaction between pp65 and cGAS was recently identified as a mechanism through which pp65 inhibits the release of cGAMP, thereby preventing STING recruitment to cGAS and impeding the expression of IFNβ [34]. STING signaling is also reported to be inhibited by pp65 via interaction with the upstream STING activator interferon gamma-inducible protein 16 (IFI16). Evidence suggests that pp65 binds to IFI16 and occludes the pyrin domains required for IFI16 oligomerization in the nucleus, preventing STING activation and anti-viral cytokine expression [35]. Contributing to this picture, pp65 has also been shown to form a complex with UL97 [36], a conserved herpesviral modulator of IFN signaling that downregulates IFN secretion via inhibition of IRF3 [37], and may have further direct or indirect effects on IFI16 activation mediated through this interaction. These recent findings underscore the extent to which pp65’s interactions with IFN signaling are continuing to be elucidated.
Evidence has emerged implicating several other tegument proteins in innate immune modulatory activities. These include the UL23, UL26, and pp71 proteins. The pp71 tegument protein, encoded by the UL82 gene, shares significant homology with its neighboring HCMV gene product pp65 [38,39] and has been identified as a key activator of the HCMV major immediate-early promoter (MIEP) [40]. Known to be required for high-titer viral replication [41], pp71 has been discovered to provide an integral defense against silencing of the viral MIEP by the host innate immune effector Daxx [42]. Through direct binding, pp71 functions to disrupt complex formation between Daxx and other transcriptional repressors such as ATRX [43], as well as target Daxx itself for proteasomal degradation [44]. Underscoring the relevance of this interaction is the observation that ablation of pp71 results in attenuated expression of HCMV immediate-early genes and impaired lytic replication, which can be rescued via inhibition of Daxx [41,45]. pp71 has also recently been found to bind STING and prevent the formation of complexes required for its translocation, sequestering it away from the nucleus [46]. Further, RNAi-mediated knockdown or gene knockout of pp71 increases host anti-viral gene expression, further emphasizing the importance of pp71 for attenuating innate immunity during infection [46].
Tegument proteins have also been shown to modulate the Type II IFN response to infection. Type II signaling, induced by IFNγ, is heavily dependent on STAT1 transcription factor homodimers that bind and activate transcription at promoters containing gamma interferon activation sequences (GAS) [47]. Recent work using UL23-deficient HCMV mutants shows that Type II IFN gene targets are upregulated during infection in the absence of UL23. These mutants are also more sensitive to challenge with IFNγ [48]. A putative binding interaction between UL23 and the STAT effector molecule N-myc interactor (NMI), identified via yeast two-hybrid screening, is hypothesized to prevent the proper activation and translocation of the STAT1 homodimers required for Type II IFN signaling [48]. These early results provide insight into HCMV’s modulation of Type II IFN, which is less well-characterized than its interplay with Type I signaling.
Complex relationships between tegument proteins and individual interferon stimulated genes are also currently being elucidated. The ubiquitin-like modifier ISG15 is a Type I IFN target that is covalently bound to target molecules, thereby altering their function (reviewed in [49]). Multiple reports have implicated ISGylation as an anti-viral defense mechanism that is triggered early during HCMV infection via cGAS-STING viral DNA sensing and restricts HCMV replication [50,51]. UL26 was recently found to interact with ISG15 as well as multiple enzymes involved in the activation and ligation of ISG15 to target proteins [51]. Interestingly, some of these ISG15 enzymes have other potentially relevant roles in the immune response outside of their ISG15-proximal functions, such as UBP43, which ligates ISG15 to target proteins but also binds IFNAR2 and occludes its interaction with JAK1 to downregulate IFN signaling [52]. During infection in the absence of immediate-early protein 1 (IE1), UL26 appears to play a role in suppressing the accumulation of ISGylated proteins [51]. Notably, UL26 itself is subject to ISG15 modification [51]; however, many questions remain about how the interactions between UL26 and the ISG15-machinery contribute to viral infection.
In addition to proteins involved in cytoplasmic DNA sensing, e.g., IFI16, ZBP1, and cGAS, the host cell also encodes proteins that sense cytoplasmic dsRNA and activate similar responses, with one of the most prominent being PKR (reviewed in [53]). PKR is an ISG with an N-terminal dsRNA-binding domain that can be activated by the presence of dsRNA as well as by a host of other stimuli including oxidative stress, cytokines, and other cellular kinases [54]. Active PKR functions by homodimerizing and autophosphorylating itself to enable a binding interaction between its C-terminal domain and the eukaryotic transcription initiation factor eIF2α, globally repressing transcription of both viral and cellular genes by interfering with the formation of the eIF2α-tRNAMet-GTP transcription initiation complex [55,56]. PKR signaling can result in a diverse array of immune outcomes including upregulated Type I IFN signaling [57] as well as increased NFκB pathway activity [58]. HCMV encodes two immediate-early gene products that specifically target PKR as a means of downregulating the IFN response and maintaining high levels of viral gene transcription during infection: IRS1 and TRS1.
Co-infection of HCMV was found to be sufficient to rescue protein translation of a Vaccinia virus mutant (VVΔE3L) that is sensitive to PKR activity [59], which ultimately led to the identification of IRS1 and TRS1 as key PKR modulatory factors [60]. Further investigation into TRS1 found that two distinct regions of this protein modulate PKR in separable fashions, with the C-terminal region capable of directly binding dsRNA and presumably preventing it from binding PKR and other dsRNA sensors, and the N-terminal region binding PKR directly and sequestering it in the nucleus to prevent its activation and downstream signaling [61,62,63]. Interestingly, both regions of TSR1 are required to fully rescue VVΔE3L-PKR related phenotypes, which include viral replication and host cell range [61], though more recent results suggest that the N-terminal region is responsible for the majority of the PKR inhibition phenotype [64].
To assess the contribution of these proteins to overall HCMV replication, mutants containing deletions of IRS1 and TRS1 both individually and in tandem were used to show that loss of either gene product in isolation did not strongly affect viral growth, but infection with an IRS1/TRS1 double deletion mutant resulted in an extreme reduction in protein synthesis and failure to replicate in primary human fibroblasts [65]. A follow up study showed that this pattern of growth correlated to levels of PKR activation as well as that viruses harboring individual mutations of IRS1 and TRS1 maintained the ability to suppress PKR activation during infection, whereas siRNA silencing of PKR rescued viral growth in the context of simultaneous loss of IRS1 and TRS1 [66]. These results indicate that modulation of IFN signaling by IRS1 and TRS1 directly contributes to infectious outcomes. As future inquiries continue to dissect the different functions of IRS1 and TRS1, it will be important to elucidate the nuances of IRS1 function, which has received less scrutiny to date than TRS1.
2.2. HCMV Immediate-Early Genes (IE) and Interferon Modulation
Immunomodulatory tegument proteins enjoy a temporal advantage over viral genes that must be expressed de novo during infection as there is no delay between the onset of infection and their time of action. However, studies investigating the role of de novo viral protein synthesis in influencing the interferon response to HCMV infection using UV-inactivated virus [30,32] and cycloheximide treatment [67] have found that, in addition to the suite of tegument-delivered viral proteins, newly synthesized viral proteins are also required for wild-type interferon pathway modulation. Recently identified as an HCMV-encoded regulator of ISG15 and ISGylation, IE1 (IE72, pUL123) was observed to strongly inhibit ISG15 transcription levels during viral infection and enable viral evasion of the ISGylation response [51]. This discovery is just the latest in a collection of work pointing to IE1’s ability to modulate Type I IFN signaling. Initial studies found that a recombinant virus lacking IE1 was vulnerable to Type I IFN treatment [68]. Further work revealed that IE1 binds to STAT2 via its carboxy-terminal acidic domain, which is also required for high-titer viral replication, and that this interaction interferes with the ability of STAT2-IFN-stimulated gene factor 3 (STAT2-ISGF3) complexes to bind to interferon stimulated response elements (ISRE) and increase the transcription of Type I IFN targets [69]. Interestingly, IE1 appears to prevent STAT2-ISGF3 complex loading onto ISRE sites without altering the abundance, phosphorylation, or complex formation ability of STAT2, relying on a mechanism of interference that has yet to be fully described [68,69]. Additionally noteworthy is that SUMOylation of IE1’s acidic domain has been shown to inhibit the IE1-STAT2 interaction and mitigate the impact of IE1 on IFN target gene expression, indicating that the cell has evolved countermeasures to respond to the activities of IE1 during infection [69].
In addition to modulation of the Type I pathway, IE1 expression has been reported to alter Type II IFN signaling. Multiple studies show that endogenous expression of IE1 in human fibroblasts is sufficient to shift the host transcriptional profile to one resembling IFNγ-treated cells [70,71] This transcriptional response was shown to occur independently of IFNγ [70], and IE1 does not appear to directly interact with STAT1, the major Type II transcription factor [72]. Further investigation led to a model in which IE1 binds STAT3 and sequesters it in the nucleus, preventing its phosphorylation [71]. In the absence of cytoplasmic STAT3, STAT1 phosphorylation and activation by the cytoplasmic kinase JAK1 is increased, resulting in higher levels of phosphorylated STAT1 localizing to the nucleus and inducing transcription of Type II IFN target genes [70]. However, other recent findings indicate that endogenous IE1 is capable of reducing STAT1 homodimer binding to Type II GAS promoter elements, complicating the story [72]. Collectively, it is clear that many questions about the interplay between IE1 and Type II IFN signaling remain to be resolved.
IE2 (IE86, pUL122) has also been closely linked with IFN modulation during infection. IFNβ production during infection is strongly inhibited by the presence of IE2 [73], which has been found to suppress IFNβ levels by binding NFκB and preventing its interaction with NFκB sites located on the IFNβ promoter, thereby inhibiting transcription [32]. Notably, a more current analysis has broadened the scope of potential interactions between IE2 and Type I IFN by showing that IE2 also targets the IFN activating molecule STING for proteasomal degradation and attenuates STING-induced transcription of IFNβ, potentially through two distinct mechanisms [74]. While many questions remain, evidence of modulation of interferon signaling by IE1 and IE2 strongly supports the notion that targeting interferon signaling at immediate-early times of infection is critical for successful HCMV infection and persistence.
2.3. HCMV-Mediated Modulation of Interferon at Later Times of Infection
The unique short region of the HCMV genome contains a series of adjacent genes with limited sequence similarity, US2–US11, that encode a set of glycoproteins implicated in a variety of activities including immune modulation [75,76]. US9 is expressed with early kinetics during viral infection and localizes to the mitochondria and endoplasmic reticulum [77], where it appears to modulate Type I IFN signaling and IFNβ production through two distinct interactions with the MAVS and STING adaptor proteins [78]. MAVS and STING both function similarly as adaptors that recruit the TBK1 kinase with its substrate, IRF3, which is then activated and localized to the nucleus to upregulate ISG transcription [78]. Expression of US9 in cells results in a strong downregulation of IRF3 activation and nuclear accumulation [78]. US9 appears to achieve this effect by damaging the cell’s mitochondria and preventing its retention of MAVS, as well as by directly binding STING and preventing its dimerization and downstream activation of IRF3 [78]. As a non-tegument HCMV protein expressed with early kinetics, US9 is of particular interest because most HCMV-encoded IFN modulatory proteins discovered to date are either delivered with the tegument or expressed immediately upon infection. In contrast, US9 is scarcely detectable within the cell prior to 6 hpi and reaches its highest level of accumulation around 48 hpi [23], appearing to be exclusively dedicated to protecting and enabling the later phases of the viral life cycle.
As discussed above, the identification of pp71 as a cGAS-STING interactor was achieved through the use of an HCMV gene expression library containing 131 constructs each encoding a unique HCMV protein [46]. Another HCMV gene product that emerged from this screen was UL31, a true-late protein required for wild-type viral growth [79,80] that was found to be capable of binding both viral DNA and host cell cGAS via its N- and C-terminal regions, respectively. UL31’s high binding affinity for cGAS, but not for DNA, suggests a non-competitive mechanism of action wherein UL31 preferentially binds cGAS in a manner that dissociates DNA from the molecule. UL31 fails to inhibit cGAMP induction of Type I ISGs but is capable of inhibiting the interferon-associated gene transcription stimulated by both HCMV infection and dsDNA, signifying that this key interaction with cGAS is critical for modulating downstream immune signaling [81]. Further, knockdown of cGAS is capable of rescuing the growth defect of UL31-deficient viral infection, indicating that the UL31/cGAS interaction has implications for overall viral growth [81]. Notably, UL31-mediated inhibition of cGAS appears to have a broad immune footprint, affecting multiple immune signaling archetypes, as infection with a UL31-deficient HCMV mutant strongly induced both ISG’s and NFκB target genes, and plasmid overexpression of UL31 in fibroblasts inhibited the activation of both ISRE-containing and NFκB reporter elements [81].
Another upstream inducer of STING, IFI16, is targeted for inhibition by the HCMV early-late gene UL97. Conserved among herpesviruses, UL97 is a viral kinase that has been shown to bind and phosphorylate IFI16, inducing its relocalization out of the nucleus of infected cells into the cytoplasm, where it is prevented from inducing an IFN response [82]. Nuclear retention of IFI16 can be rescued by treatment with an inhibitor that reduces UL97 kinase function or by removing the UL97 reading frame from the HCMV genome, further establishing this link between UL97 and IFI16 export [82].
2.4. Modulation of Interferon during Latency
Work using an IRS1 and TRS1 double mutant (ΔIRS1/ΔTRS1) to investigate PKR modulation during infection was instrumental in uncovering another HCMV gene product that alters the host cell’s ability to sense viral dsRNA: ORF94 (UL126a). Oligoadenylate synthetase (OAS) proteins are dsRNA-binding enzymes that activate cellular RNases in response to dsRNA detection, degrading host and viral RNA and down regulating overall rates of protein synthesis (reviewed in [83]). HCMV infection institutes a block to OAS signaling, and it was observed that while HCMV-encoded TRS1 and IRS1 are capable of downregulating this pathway in certain contexts [60], a mutant HCMV lacking both of these open reading frames still inhibited OAS activation [65]. HCMV ORF94 was identified as a potential candidate that may be mediating this OAS block by inhibiting the expression of OAS1 in multiple contexts, including during productive infection and in the face of interferon stimulation [84]. Notably, HCMV ORF94 is a latency-associated ORF encoded by transcripts expressed during latent infection [85]. While extremely intriguing, it still remains to be seen how ORF94 and its modulation of the IFN response contributes to latency establishment, maintenance, or reactivation.
3. HCMV Modulation of NFκB Signaling
The NFκB signaling network regulates a wide variety of pro-inflammatory processes that ultimately shape innate and adaptive immune responses via transcriptional regulation of numerous NFκB responsive genes. NFκB signaling can be activated by a myriad of inducers including infectious agents, paracrine signaling factors, or environmental stress [86]. Highlighting its importance as a defense against infection, a wide variety of evolutionarily diverse viruses including Human Immunodeficiency Virus-1 (HIV-1), HCMV, Herpes Simplex Virus-1 (HSV-1), and Epstein Barr Virus (EBV) have evolved mechanisms to modulate NFκB signaling [87]. Numerous interactions between HCMV and NFκB signaling have been described over the years (Figure 2, and reviewed in [87,88]). HCMV gene products have been shown to both inhibit NFκB signaling and activate facets of the NFκB pathway to support lytic replication or induce reactivation from latency [89,90], suggesting a nuanced relationship. This highlights the reality that NFκB signaling is not simply binary, but rather that NFκB endpoint signaling is capable of multiple distinct transcriptional landscapes depending on specific upstream stimuli. The relationship between HCMV and NFκB is shaped by multiple HCMV gene products, including both proteins and miRNAs, which serve to modulate various aspects of NFκB signaling. Less clear are how the various facets of HCMV-mediated modulation of NFκB contribute to the variety of biological contexts of HCMV infection, including viral persistence, shedding, latency, reactivation, tropism, and pathogenesis.
Most effectors of the NFκB response converge upon the activation of a serine-specific IκB kinase (IKK) complex comprised of different combinations of three distinct subunits: IKKα, IKKβ and IKKγ/NEMO (Figure 2). Canonical NFκB signaling relies on a tripartite complex consisting of one of each IKK subunit, while non-canonical NFκB functions through an IKKα dimer [86]. The mechanisms through which the cytoplasmic NFκB subunit complexes are activated represent a major difference between these two sub-pathways (Figure 2). In canonical NFκB signaling, an IKK complex containing NEMO phosphorylates the repressor protein IκB, marking it for ubiquitination and degradation. IκB represses the canonical NFκB transcription factors p50 and p65 (RelA) in the cytoplasm, which are freed upon IκB phosphorylation/degradation, resulting in their nuclear translocation and subsequent transcriptional activation of NFκB targets [80]. During non-canonical NFκB signaling an IKKα homodimer acts as the kinase that phosphorylates the C-terminus of p100, which resides in an inhibitory complex with RelB in the cytoplasm. This phosphorylation of p100 results in its processing to p52, which in complex with RelB, can enter the nucleus to modulate NFκB target transcription [91] (Figure 2).
During HCMV infection of fibroblasts NFκB activation appears to follow a specific sequence in which the pathway is active early in infection, but is then repressed from middle to late time points of the viral life cycle. At the earliest time of infection, i.e. envelope fusion, the HCMV glycoproteins B and H (gB/gH), encoded by UL55 and UL75, respectively, bind to Toll-like receptor 2 (TLR-2) on the surface of the cell and induce a canonical NFκB signaling cascade resulting in the excretion of pro-inflammatory and anti-viral cytokines [92]. This immediate enhancement of NFκB activation appears to be pro-viral, as HCMV’s MIEP possesses NFκB binding motifs [93] that facilitate the expression of IE genes, an effect that can be enhanced by TNFα stimulation [94]. Further evidence supporting a pro-viral aspect of early NFκB activation for infection include the findings that dominant-negative constructs targeting key NFκB constituents such as IKKα, IKKβ, and IκBα reduce MIEP activation [95]. These findings suggest that NFκB activation at early times is important for optimal transactivation of the MIEP. However, the utilization of HCMV mutants lacking the NFκB motifs in the MIEP did not result in significant attenuation of infection or IE gene product accumulation [96], suggesting that additional host transcription factor binding motifs present in the MIEP, including CREB (cAMP response element binding) and ATF (activating transcription factor) sites [97], may be sufficient to activate IE gene transcription even in the absence of p65/p50 binding. This remains an unresolved issue, and further inquiries have suggested that this nuanced interaction between viral transcription and NFκB signaling can be influenced by numerous factors including the host cell’s progression through the cell cycle [89], the adaptation of HCMV strains to laboratory passage conditions [98], and host cell lineage [99].
3.1. HCMV Tegument Proteins and NFκB Modulation
Concurrent with envelope fusion, the virion releases viral tegument proteins into the cytoplasm which disseminate and begin to modulate a number of cellular pathways. The pp65 protein, as discussed above, likely plays a role in blocking the host IFN response during early infection that is not yet fully understood. In addition to this IFN modulatory role, infection with a pp65-deficient mutant HCMV increases the accumulation of NFκB target genes and induces the nuclear binding activity of NFκB transcription factors [31], suggesting an important contribution to NFκB regulation. Mechanistically, however, it is unclear how pp65 modulates NFκB activity. Further, the extent to which pp65’s modulation of IFN and NFκB signaling might be functionally related is uncertain.
The UL26 protein is also delivered with the tegument and has been shown to antagonize NFκB activity. A UL26 deletion mutant virus is severely attenuated, and UL26 has been shown to be necessary to inhibit IKK complex phosphorylation and RelB translocation during infection [100,101]. Further, expression of UL26 by itself is sufficient to block TNFα-mediated NFκB activation [100,101]. However, despite its presence in the tegument, UL26 does not block the activation of NFκB at the earliest times of infection. UL26 is thought to have a stronger role in the attenuation of NFκB activity that occurs later during infection, when UL26 localizes to the cytoplasm, as opposed to early times when it localizes in the nucleus [102]. However, the possibility of an interaction between UL26 and NFκB at early times during infection cannot be ruled out, as UL26-deficient virus is more sensitive to challenge with TNFα [100]. Notably, UL26 is capable of inhibiting IKK complex activation in the face of diverse upstream stimuli including TNFα signaling and Sendai virus infection, suggesting that it is acting at the level of the IKK complex, where these signaling cascades converge [100], but the exact mechanism of UL26’s inhibition of NFκB signaling remains to be elucidated
HCMV tegument proteins are also capable of inducing pro-viral NFκB signaling. UL76, a tegument-associated endonuclease, is reported to activate the canonical NFκB pathway through the DNA damage response and induce IL-8 production [103]. This UL76-mediated increase in IL-8 production was shown to be dependent on the cellular kinases Ataxia-telangiectasia mutated (ATM) and IKKβ; however, ablation of ATM expression in cells or of the key endonuclease motif amino acids present in UL76 failed to completely restore IL-8 production back to wild-type levels during infection [103], implying that additional aspects of NFκB signaling might be contributing to this phenotype. A UL76 deletion mutant has a significant growth defect [104], but the contributions of increased IL-8 production to this attenuation are not clear.
HCMV virions have been found to incorporate cellular mRNAs and proteins [27,105]. This raises the possibility that, in addition to viral factors, virion-associated cellular factors could also be modulating NFκB signaling. One such example is the virion packaging of casein kinase II (CKII), which has been found in the virion tegument and is reported to activate NFκB signaling through phosphorylation of the IκB repressor, thereby releasing the associated NFκB subunits to localize to the nucleus and induce NFκB-dependent transcription [106]. The extent to which cellularly-derived, virion-associated NFκB modulators contribute to the various facets of HCMV infection is still largely not known.
3.2. HCMV Immediate-Early Genes and NFκB Modulation
The IE proteins expressed upon MIEP stimulation interact with the NFκB pathway in diverse ways. IE1, a promiscuous transactivator of NFκB pathway constituents and their downstream targets during infection, has been implicated in the upregulation of p65, IL-6, TNF-α and IL-8 as well as the induction of p52/RelB heterodimer binding activity in the nucleus [88]. At immediate-early times, the virus also produces UL144, a TNF-receptor-like transmembrane receptor with immediate-early expression kinetics [107], which activates expression of the immune cytokine CCL22 [108]. Mechanistically, UL144 complexes with TRAF6 in perinuclear regions of the cell to enable NFκB transcription factor translocation and binding, and siRNA targeting UL144, TRAF6, or NFκB all ablate the downstream CCL22 expression induced by infection [108]. The CCL22 cytokine has been noted to play a chemoattractant role in recruiting Th2 and regulatory T-cells (Tregs) to mediate adaptive immune responses [108]. The UL144 open reading frame is lost in extensively laboratory passaged strains of HCMV, potentially due to its activation of NFκB signaling, which could be a detriment to viral fitness during in vitro fibroblast infection [109,110].
In addition to immediate-early expression of the NFκB agonists IE1 and UL144, IE2 inhibits host NFκB signaling at all points during HCMV infection through a still-controversial model: either by blocking NFκB subunit dimer interactions or preventing subunit interactions with specific NFκB target promoters, e.g., IL-6 [32,111]. Interestingly, the antagonistic effects of IE2 do not prevent UL144 from inducing NFκB [112], which highlights the specificity with which HCMV is able to tailor NFκB signaling. Collectively the data suggest that at early points of infection the virus seems to be operating within an optimal pro-inflammatory signaling window, with just enough NFκB transcription factor binding to transactivate the viral MIEP, but still staying below a threshold that might trigger a broader anti-viral immune response.
3.3. HCMV-Mediated Modulation of NFκB at Later Times of Infection
At some point during the transition from the immediate-early stages of infection, where active NFκB signaling is observed, to the later stages of infection, the viral actions towards NFκB become much more inhibitory. This transition coincides with the increased expression of multiple HCMV genes that antagonize NFκB activity. One such gene is UL111a, also known as cmvIL-10 for its functional similarity to the cellular cytokine IL-10. cmvIL-10 shares only approximately 27% homology with host IL-10, but binds as a homodimer to the human IL-10 receptor and blocks NFκB signaling by preventing IκBα degradation in a similar manner to IL-10 [113,114,115]. In addition, cmvIL-10 also has a significant immunosuppressive effect on interferon signaling. In peripheral blood mononuclear cells (PBMCs), TLR-agonist treated media from wild-type AD169 infection is sufficient to inhibit the production of IFNα when compared to similarly treated media from a cmvIL-10 knockout virus [116]. Further, cmvIL-10 alone is sufficient to do the same in plasmacytoid DCs [117]. The mechanism behind these inhibitions appears to involve activation of STAT3 [118], but is still poorly understood. It remains unknown if the suppressive effects of cmvIL-10 on IFN and NFκB signaling are separable. Studies of the cmvIL-10/interferon signaling axis suggest that the molecule may be acting in a paracrine manner to activate defenses in uninfected cells, which could represent a promising avenue of further inquiry.
3.4. HCMV miRNAs and NFκB Modulation
Viral miRNAs represent additional tools that HCMV employs to undermine host cell defenses at later points of infection. HCMV encodes 26 miRNAs, each approximately 22nt in length, that have been implicated in modulating a wide variety of cellular pathways and processes including vesicle transport, cytokine secretion, immune signaling, and progression through the cell cycle, (reviewed in [119] and [120]). Throughout the course of infection, beginning with immediate-early gene expression, HCMV miRNAs begin to accumulate, becoming abundant by late infection [119,121]. HCMV miR-US5-1 and miRUL112-3p have been shown to prevent NFκB cytokine signaling by specifically downregulating the expression of the key kinases IKKα and IKKβ [122]. In addition to blocking the NFκB signal relay set off by cytokine detection, miR-US5-2 is able to block the infected cells’ secretion of cytokines [123], thereby ceasing the positive feedback loop of NFκB activation and ultimately returning the pathway to its initial inactive state. During latent infection, miR-UL148D is one of the most highly expressed miRNAs, and has been demonstrated to block NFκB upstream adapters and repress IL-6 production [124], thus allowing the infected cell to evade host immunity.
3.5. Latency-Associated NFκB Modulators
A hallmark of all herpesviruses is the ability to enter latency, persisting with limited lytic viral replication in the face of a primed immune response and capable of reactivation during times of stress or immunosuppression, resulting in viral dissemination and potential pathologies. The signals that reactivate HCMV from latency remain incompletely understood, but immunosuppression and inflammation are thought to play major roles (reviewed in [125]). Consistent with this view, reports indicate that viral genes activate NFκB during reactivation [126], and, further, NFκB activation has been linked to HCMV reactivation via NFκB subunit enhancement of MIEP expression [127,128]. The viral chemokine receptor US28, expressed with early kinetics during lytic infection, is one of the few viral proteins expressed during latency as demonstrated in latently infected THP-1 monocytes [128]. US28 has been implicated in activating the MIEP through NFκB signaling [126]; it is possible that during latent expression, US28 activates the MIEP and aids in reactivation from latency. US28 induces constitutive NFκB activation through its interaction with the Gq/11 G protein, which mediates the release of Gβγ subunits that induce downstream NFκB activity [129]. Although in general US28 has appeared to stimulate NFκB activity, recent work suggests that US28 attenuates multiple cell signaling pathways including NFκB, which is required to maintain latency as mutants lacking US28 return to their lytic phase and infected cells are subsequently targeted by T-cells [130]. It is clear that US28 plays a complicated role in the HCMV life cycle; and, like other viral factors, possesses more than one role that may seem counterintuitive, but that are likely important in different infectious contexts.
Another viral protein expressed during latency, UL138, acts by activating and stabilizing the cell surface expression of TNFR1 [131,132,133]. Interestingly, while UL138 appears to promote the sensitivity of latently infected cells to TNFα, reporter assays show that UL138 strongly represses MIEP transactivation [134] and ChIP (chromatin-immunoprecipitation) assays suggest it prevents cellular demethylases from interacting with the MIEP [135], leading to the conclusion that this protein is also playing a central role in maintaining HCMV’s latent state. These results represent some of the first forays into exploring the immunomodulatory potential of HCMV latency-associated genes, and the data suggest that there is a complex interplay between pro- and anti-viral manipulations of immune signaling as the virus maintains latent infection despite silencing much of its transcription.
4. Conclusions and Future Perspectives
Reports over the past 25 years have made it abundantly clear that HCMV devotes substantial genetic resources towards manipulation of interferon and NFκB signaling. The list of involved genes is large and still growing, with a variety of HCMV gene products playing modulatory roles in various contexts. While a multitude of questions remain, certain themes and patterns have emerged. For example, with respect to IFN signaling, while some ISGs such as viperin and Cox2 (reviewed in [12]) appear to be co-opted by the virus for pro-viral activities, almost all HCMV gene products that have been identified to play a role in IFN signaling serve to attenuate this host cell response to support robust infection, suggesting that HCMV goes to great lengths to inhibit IFN signaling to enable infection.
In contrast, the literature regarding HCMV’s modulation of NFκB suggests a more complex picture. NFκB activation during lytic infection of fibroblasts appears to be biphasic with a pro-inflammatory, pro-viral NFκB signaling environment instituted during the earliest times of infection, which subsequently shifts to broadly inhibitory of NFκB at later time points of infection. As discussed above, current data suggest a model in which early NFκB activation supports infection through increased MIEP transcription, whereas later NFκB inhibition prevents the secretion of NFκB-regulated anti-viral factors, e.g., cytokines. However, many questions remain. For one, the sheer number of HCMV gene products that modulate NFκB inflammatory signaling in diverse ways suggests a more nuanced story. It seems likely that these NFκB modulatory gene products work in different infectious contexts to support varied aspects of viral infection, including robust lytic infection, the establishment of latency, and reactivation from the latent state. In this regard, the activities of NFκB-modulatory viral proteins will likely be sensitive to a variety of cellular states including cell type, differentiation status, and the inflammatory environment.
A typical in vivo infection progresses from the mucosal epithelial cells to responding immune cells, with subsequent seeding of lymph nodes and infection of progenitor cells that will serve as latency reservoirs, followed by some level of either subclinical or pathogenic reactivation back in the mucosal epithelia. We propose that cell-type and context specific HCMV-mediated modulation of NFκB will be critical at each step of this process. Furthering our understanding of how the relationship between HCMV and NFκB shapes viral biology and pathogenesis will require elucidating how specific mechanisms of HCMV-mediated modulation of NFκB contribute to key events during the in vivo viral life cycle in physiologically relevant cell types. While this will be challenging given the limitations of our current in vivo and in vitro models, recent developments in humanized mice, as well as explant and organoid culture techniques will yield exciting, novel opportunities to address these issues. The field has made substantial progress, likely identifying the majority of the viral players involved in NFκB modulation. It is time to turn our attention to identifying how these viral NFκB modulators contribute to various infectious contexts, and to developing the tools and experimental systems required to do so. Given the importance of these interactions to infectious outcomes, further elucidating how they contribute to infection will likely provide novel avenues to limit HCMV-associated pathogenesis.
Author Contributions
Conceptualization and Writing, C.M.G., J.H.C., and J.M.
Funding
C.M.G. was supported by an NIH training grant from the Training Program in Oral Science (T90-DE021985-07). The work was also supported by an NIH grant AI127370 to J.M. and by a Research Scholar Grant from the American Cancer Society (RSG-15-049-01-MPC).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gerna, G.; Baldanti, F.; Revello, M.G. Pathogenesis of human cytomegalovirus infection and cellular targets. Hum. Immunol. 2004, 65, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Pass, R.F. Cytomegalovirus. In Fields’ Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Williams and Wilkins: New York, NY, USA, 2001; pp. 2675–2705. [Google Scholar]
- Neiman, P.; Wasserman, P.B.; Wentworth, B.B.; Kao, G.F.; Lerner, K.G.; Storb, R.; Buckner, C.D.; Clift, R.A.; Fefer, A.; Fass, L.; et al. Interstitial pneumonia and cytomegalovirus infection as complications of human marrow transplantation. Transplantation 1973, 15, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Pinninti, S.G.; Ross, S.A.; Shimamura, M.; Novak, Z.; Palmer, A.L.; Ahmed, A.; Tolan, R.W., Jr.; Bernstein, D.I.; Michaels, M.G.; Sánchez, P.J. Comparison of saliva PCR assay versus rapid culture for detection of congenital cytomegalovirus infection. Pediatr. Infect. Dis. J. 2015, 34, 536–537. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Ross, S.A.; Shimamura, M.; Palmer, A.L.; Ahmed, A.; Michaels, M.G.; Sánchez, P.J.; Bernstein, D.I.; Tolan, R.W., Jr.; Novak, Z. Saliva polymerase-chain-reaction assay for cytomegalovirus screening in newborns. N. Engl. J. Med. 2011, 364, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Wolff, T. Activation of the antiviral kinase PKR and viral countermeasures. Viruses 2009, 1, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARS) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Servant, M.J.; Hiscott, J. The interferon antiviral response: From viral invasion to evasion. Curr. Opin. Infect. Dis. 2002, 15, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.R.; Ashkar, A.A.; Mossman, K.L. The nitric oxide pathway provides innate antiviral protection in conjunction with the type I interferon pathway in fibroblasts. PLoS ONE 2012, 7, e31688. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A.; Hauka, S.; Maywald, M.; Le, V.T.K.; Schmidt, S.K.; Däubener, W.; Hengel, H. Checks and balances between human cytomegalovirus replication and indoleamine-2,3-dioxygenase. J. Gen. Virol. 2014, 95, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Bekisz, J.; Schmeisser, H.; Hernandez, J.; Goldman, N.D.; Zoon, K.C. Mini review Human interferons alpha, beta and omega. Growth Factors 2004, 22, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Amsler, L.; Verweij, M.C.; DeFilippis, V.R. The tiers and dimensions of evasion of the type I interferon response by human cytomegalovirus. J. Mol. Biol. 2013, 425, 4857–4871. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Prasanna, S.J.; Chandrasekar, B.; Nandi, D. Gene modulation and immunoregulatory roles of interferonγ. Cytokine 2010, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Cong, J.-P.; Shenk, T. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAS: Induction of interferon-responsive RNAS. Proc. Natl. Acad. Sci. USA 1997, 94, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, V.R.; Alvarado, D.; Sali, T.; Rothenburg, S.; Früh, K. Human cytomegalovirus induces the interferon response via the DNA sensor zbp1. J. Virol. 2010, 84, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Paijo, J.; Döring, M.; Spanier, J.; Grabski, E.; Nooruzzaman, M.; Schmidt, T.; Witte, G.; Messerle, M.; Hornung, V.; Kaever, V. Cgas senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog. 2016, 12, e1005546. [Google Scholar] [CrossRef] [PubMed]
- Le-Trilling, V.; Trilling, M. Attack, parry and riposte: Molecular fencing between the innate immune system and human herpesviruses. Tissue Antigens 2015, 86, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Trilling, M.; Le, V.T.K.; Hengel, H. Interplay between cmvs and interferon signaling: Implications for pathogenesis and therapeutic intervention. Future Microbiol. 2012, 7, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Tomasec, P.; Huttlin, E.L.; Fielding, C.A.; Nusinow, D.; Stanton, R.J.; Wang, E.C.; Aicheler, R.; Murrell, I.; Wilkinson, G.W. Quantitative temporal viromics: An approach to investigate host-pathogen interaction. Cell 2014, 157, 1460–1472. [Google Scholar] [CrossRef] [PubMed]
- Le, V.T.K.; Trilling, M.; Wilborn, M.; Hengel, H.; Zimmermann, A. Human cytomegalovirus interferes with signal transducer and activator of transcription (STAT) 2 protein stability and tyrosine phosphorylation. J. Gen. Virol. 2008, 89, 2416–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.M.; Zhang, Y.; Rahill, B.M.; Waldman, W.J.; Sedmak, D.D. Human cytomegalovirus inhibits IFN-α-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-α signal transduction. J. Immunol. 1999, 162, 6107–6113. [Google Scholar] [PubMed]
- Miller, D.M.; Rahill, B.M.; Boss, J.M.; Lairmore, M.D.; Durbin, J.E.; Waldman, J.W.; Sedmak, D.D. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the JAK/STAT pathway. J. Exp. Med. 1998, 187, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Paša-Tolić, L.; Wang, D.; Camp, D.G.; Rodland, K.; Wiley, S. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.J.; Riddell, S.R.; Plachter, B.; Greenberg, P.D. Cytomegalovirus selectively blocks antigen processing and presentation of its immediate—Early gene product. Nature 1996, 383, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Arnon, T.I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 2005, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Abate, D.A.; Watanabe, S.; Mocarski, E.S. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 2004, 78, 10995–11006. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Shenk, T. Human cytomegalovirus ul83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11439–11444. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.T.; Bresnahan, W.A. Human cytomegalovirus immediate—Early 2 protein IE86 blocks virus-induced chemokine expression. J. Virol. 2006, 80, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Damania, B. The CGAS-sting defense pathway and its counteraction by viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; de Andrea, M. Human cytomegalovirus tegument protein pp65 (pUL83) dampens type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J. Virol. 2018, 92, e01774-01717. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kamil, J.P.; Coen, D.M. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J. Virol. 2007, 81, 10659–10668. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, K.S.; Flano, E.; Wu, T.-T.; Tong, L.M.; Park, A.N.; Song, M.J.; Sanchez, D.J.; O’Connell, R.M.; Cheng, G. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 2009, 5, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Nowak, B.; Gmeiner, A.; Sarnow, P.; Levine, A.J.; Fleckenstein, B. Physical mapping of human cytomegalovirus genes: Identification of DNA sequences coding for a virion phosphoprotein of 71 kDa and a viral 65-kDa polypeptide. Virology 1984, 134, 91–102. [Google Scholar] [CrossRef]
- Rüger, B.; Klages, S.; Walla, B.; Albrecht, J.; Fleckenstein, B.; Tomlinson, P.; Barrell, B. Primary structure and transcription of the genes coding for the two virion phosphoproteins pp65 and pp71 of human cytomegalovirus. J. Virol. 1987, 61, 446–453. [Google Scholar] [PubMed]
- Liu, B.; Stinski, M.F. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J. Virol. 1992, 66, 4434–4444. [Google Scholar] [PubMed]
- Bresnahan, W.A.; Shenk, T.E. Ul82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14506–14511. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, S.R.; Bresnahan, W.A. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 2006, 80, 6188–6191. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, V.; McFarlane, S.; Everett, R.D.; Preston, C.M. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008, 82, 12543–12554. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 2007, 367, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavalai, N.; Papior, P.; Rechter, S.; Stamminger, T. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 2008, 82, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Su, S.; Gao, Y.-Q.; Wang, P.-P.; Huang, Z.-F.; Hu, M.-M.; Luo, W.-W.; Li, S.; Luo, M.-H.; Wang, Y.-Y. Human cytomegalovirus tegument protein UL82 inhibits sting-mediated signaling to evade antiviral immunity. Cell Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Sheng, J.; Vu, G.-P.; Liu, Y.; Foo, C.; Wu, S.; Trang, P.; Paliza-Carre, M.; Ran, Y.; Yang, X. Human cytomegalovirus UL23 inhibits transcription of interferon-γ stimulated genes and blocks antiviral interferon-γ responses by interacting with human n-myc interactor protein. PLoS Pathog. 2018, 14, e1006867. [Google Scholar] [CrossRef] [PubMed]
- Perng, Y.-C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Mohr, I. Restriction of human cytomegalovirus replication by ISG15, a host effector regulated by cGAS-STING double-stranded-DNA sensing. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, E.T.; Kim, Y.E.; Lee, M.K.; Kwon, K.M.; Kim, K.I.; Stamminger, T.; Ahn, J.H. Consecutive inhibition of ISG15 expression and isgylation by cytomegalovirus regulators. PLoS Pathog. 2016, 12, e1005850. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.S.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, M.; Berg, M. The multiple faces of proteinkinase r in antiviral defense. Virulence 2013, 4, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Chong, K.; Kumar, A.; Williams, B. Identification of double-stranded RNA-binding domains in the interferon-induced double-stranded RNA-activated p68 kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 5447–5451. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Frank, J.; Kinzy, T.; Mathews, M.; Sonenberg, N.; Hershey, J. Translational control in biology and medicine. In Structure and Function of the Eukaryotic Ribosome and Elongation Factors; Mathews, M.B., Sonenberg, N., Hershey, J.W.B., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2007; pp. 71–72. [Google Scholar]
- Schulz, O.; Pichlmair, A.; Rehwinkel, J.; Rogers, N.C.; Scheuner, D.; Kato, H.; Takeuchi, O.; Akira, S.; Kaufman, R.J.; Sousa, C.R. Protein kinase r contributes to IFN-α/β production during viral infection by regulating IFN mRNA integrity. Cell Host Microbe 2010, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-κB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-κB -inducing kinase and IκB kinase. Mol. Cell. Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Jarrahian, S.; Harper, V.M.; Geballe, A.P. Complementation of vaccinia virus lacking the double-stranded RNA-binding protein gene E3L by human cytomegalovirus. J. Virol. 2002, 76, 4912–4918. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Hakki, M.; de Niro, K.L.; Geballe, A.P. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 2004, 78, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Hakki, M.; Geballe, A.P. Double-stranded RNA binding by human cytomegalovirus PTRS1. J. Virol. 2005, 79, 7311–7318. [Google Scholar] [CrossRef] [PubMed]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [PubMed]
- Bierle, C.J.; Semmens, K.M.; Geballe, A.P. Double-stranded RNA binding by the human cytomegalovirus PKR antagonist TRS1. Virology 2013, 442, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Vincent, H.A.; Ziehr, B.; Moorman, N.J. Mechanism of protein kinase R inhibition by human cytomegalovirus pTRS1. J. Virol. 2017, 91, e01574-16. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Ziehr, B.; Vincent, H.A.; Moorman, N.J. Human cytomegalovirus pTRS1 and pIRS1 antagonize protein kinase r to facilitate virus replication. J. Virol. 2016, 90, 3839–3848. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Wing, B.; Coleman, D.; Shenk, T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: Viral block to the accumulation of antiviral mRNAs. J. Virol. 2001, 75, 12319–12330. [Google Scholar] [CrossRef] [PubMed]
- Paulus, C.; Krauss, S.; Nevels, M. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 3840–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, Y.H.; Kim, Y.E.; Kim, E.T.; Park, J.J.; Song, M.J.; Zhu, H.; Hayward, G.S.; Ahn, J.-H. Binding STAT2 by the acidic domain of human cytomegalovirus IE1 promotes viral growth and is negatively regulated by SUMO. J. Virol. 2008, 82, 10444–10454. [Google Scholar] [CrossRef] [PubMed]
- Knoblach, T.; Grandel, B.; Seiler, J.; Nevels, M.; Paulus, C. Human cytomegalovirus IE1 protein elicits a type II interferon-like host cell response that depends on activated stat1 but not interferon-γ. PLoS Pathog. 2011, 7, e1002016. [Google Scholar] [CrossRef] [PubMed]
- Harwardt, T.; Lukas, S.; Zenger, M.; Reitberger, T.; Danzer, D.; Übner, T.; Munday, D.C.; Nevels, M.; Paulus, C. Human cytomegalovirus immediate-early 1 protein rewires upstream STAT3 to downstream stat1 signaling switching an il6-type to an IFNΓ-like response. PLoS Pathog. 2016, 12, e1005748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, B.; Cook, C.H.; Trgovcich, J. The carboxy terminal region of the human cytomegalovirus immediate early 1 (IE1) protein disrupts type ii inteferon signaling. Viruses 2014, 6, 1502–1524. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.T.; Bresnahan, W.A. Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced β interferon production. J. Virol. 2005, 79, 3873–3877. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Kim, Y.-E.; Stinski, M.F.; Ahn, J.-H.; Song, Y.-J. Human cytomegalovirus IE2 86 kDa protein induces sting degradation and inhibits cGAMP-mediated IFN-β induction. Front. Microbiol. 2017, 8, 1854. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.R.; Muzithras, V.P. A cluster of dispensable genes within the human cytomegalovirus genome short component: IRS1, US1 through US5, and the US6 family. J. Virol. 1992, 66, 2541–2546. [Google Scholar] [PubMed]
- Huber, M.T.; Tomazin, R.; Wisner, T.; Boname, J.; Johnson, D.C. Human cytomegalovirus US7, US8, US9, and US10 are cytoplasmic glycoproteins, not found at cell surfaces, and US9 does not mediate cell-to-cell spread. J. Virol. 2002, 76, 5748–5758. [Google Scholar] [CrossRef] [PubMed]
- Mandic, L.; Miller, M.S.; Coulter, C.; Munshaw, B.; Hertel, L. Human cytomegalovirus US9 protein contains an N-terminal signal sequence and a C-terminal mitochondrial localization domain, and does not alter cellular sensitivity to apoptosis. J. Gen. Virol. 2009, 90, 1172–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian Choi, H.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westdorp, K.N.; Sand, A.; Moorman, N.J.; Terhune, S.S. Cytomegalovirus late protein pUL31 alters Pre-rRNA expression and nuclear organization during infection. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.-F.; Zou, H.-M.; Liao, B.-W.; Zhang, H.-Y.; Yang, Y.; Fu, Y.-Z.; Wang, S.-Y.; Luo, M.-H.; Wang, Y.-Y. Human cytomegalovirus protein UL31 inhibits DNA sensing of CGAS to mediate immune evasion. Cell Host Microbe 2018, 24, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Dell’Oste, V.; Gatti, D.; Gugliesi, F.; de Andrea, M.; Bawadekar, M.; Cigno, I.L.; Biolatti, M.; Vallino, M.; Marschall, M.; Gariglio, M. Innate nuclear sensor ifi16 translocates into the cytoplasm during early stage of in vitro HCMV infection and is entrapped in the egressing virions during late stage. J. Virol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.Y.; Kang, J.-S.; Hwang, Y.S.; Kim, Y.-J. Oligoadenylate synthase-like (OASL) proteins: Dual functions and associations with diseases. Exp. Mol. Med. 2015, 47, e144. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.C.; Avdic, S.; Cao, J.Z.; Mocarski, E.S.; White, K.L.; Abendroth, A.; Slobedman, B. Inhibition of 2′, 5′-oligoadenylate synthetase expression and function by the human cytomegalovirus ORF94 gene product. J. Virol. 2011, 85, 5696–5700. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Xu, J.; Mocarski, E.S. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 1996, 93, 11137–11142. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.G. Nf-κb transcription factors. Boston University.
- Hiscott, J.; Kwon, H.; Génin, P. Hostile takeovers: Viral appropriation of the NF-κB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.H.; Nelson, J.A. Modulation of the NF-κB signalling pathway by human cytomegalovirus. Virology (Hyderabad) 2017, 1, 104. [Google Scholar] [PubMed]
- Caposio, P.; Luganini, A.; Hahn, G.; Landolfo, S.; Gribaudo, G. Activation of the virus-induced IKK/ NF-κB signalling axis is critical for the replication of human cytomegalovirus in quiescent cells. Cell. Microbiol. 2007, 9, 2040–2054. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynagh, P.N. The NF-κB pathway. J. Cell Sci. 2005, 118, 4589–4592. [Google Scholar] [CrossRef] [PubMed]
- Yurochko, A.D.; Hwang, E.S.; Rasmussen, L.; Keay, S.; Pereira, L.; Huang, E.S. The human cytomegalovirus UL55 (gB) and UL 75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-κB during infection. J. Virol. 1997, 71, 5051–5059. [Google Scholar] [PubMed]
- Cherrington, J.M.; Mocarski, E.S. Human cytomegalovirus ie1 transactivates the α promoter-enhancer via an 18-base-pair repeat element. J. Virol. 1989, 63, 1435–1440. [Google Scholar] [PubMed]
- Prösch, S.; Staak, K.; Stein, J.; Liebenthal, C.; Stamminger, T.; Volk, H.-D.; Krüger, D.H. Stimulation of the human cytomegalovirus ie enhancer/promoter in hl-60 cells by tnfα is mediated via induction of NF-κB. Virology 1995, 208, 197–206. [Google Scholar] [CrossRef] [PubMed]
- DeMeritt, I.B.; Milford, L.E.; Yurochko, A.D. Activation of the NF-κB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J. Virol. 2004, 78, 4498–4507. [Google Scholar] [CrossRef] [PubMed]
- Gustems, M.; Borst, E.; Benedict, C.A.; Pérez, C.; Messerle, M.; Ghazal, P.; Angulo, A. Regulation of the transcription and replication cycle of human cytomegalovirus is insensitive to genetic elimination of the cognate NF-κB binding sites in the enhancer. J. Virol. 2006, 80, 9899–9904. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Shenk, T.; Pass, R.F. Cytomegalovirus. In Fields Virology, 5th ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2006; Volume 1. [Google Scholar]
- Ho, C.M.; I’ah, Z.; Tan, L.; Zhang, T.; Gray, N.S.; Strang, B.L. Inhibition of IKK α by bay61-3606 reveals IKK α-dependent histone h3 phosphorylation in human cytomegalovirus infected cells. PLoS ONE 2016, 11, e0150339. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Coaquette, A.; Davrinche, C.; Herbein, G. Bcl-3-regulated transcription from major immediate-early promoter of human cytomegalovirus in monocyte-derived macrophages. J. Immunol. 2009, 182, 7784–7794. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.; Schafer, X.; Martinez-Sobrido, L.; Munger, J. The human cytomegalovirus UL26 protein antagonizes NF-κB activation. J. Virol. 2014, 88, 14289–14300. [Google Scholar] [CrossRef] [PubMed]
- Lorz, K.; Hofmann, H.; Berndt, A.; Tavalai, N.; Mueller, R.; Schlotzer-Schrehardt, U.; Stamminger, T. Deletion of open reading frame UL26 from the human cytomegalovirus genome results in reduced viral growth, which involves impaired stability of viral particles. J. Virol. 2006, 80, 5423–5434. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Yu, D.; Shenk, T. UL26-deficient human cytomegalovirus produces virions with hypophosphorylated pp28 tegument protein that is unstable within newly infected cells. J. Virol. 2006, 80, 3541–3548. [Google Scholar] [CrossRef] [PubMed]
- Costa, H.; Nascimento, R.; Sinclair, J.; Parkhouse, R.M.E. Human cytomegalovirus gene UL76 induces IL-8 expression through activation of the DNA damage response. PLoS Pathog. 2013, 9, e1003609. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terhune, S.S.; Schroer, J.; Shenk, T. RNAs are packaged into human cytomegalovirus virions in proportion to their intracellular concentration. J. Virol. 2004, 78, 10390–10398. [Google Scholar] [CrossRef] [PubMed]
- Nogalski, M.T.; Podduturi, J.P.; DeMeritt, I.B.; Milford, L.E.; Yurochko, A.D. The human cytomegalovirus virion possesses an activated casein kinase II that allows for the rapid phosphorylation of the inhibitor of NF-κB, IκBα. J. Virol. 2007, 81, 5305–5314. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.A.; Butrovich, K.D.; Lurain, N.S.; Corbeil, J.; Rooney, I.; Schneider, P.; Tschopp, J.; Ware, C.F. Cutting edge: A novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J. Immunol. 1999, 162, 6967–6970. [Google Scholar] [PubMed]
- Poole, E.; King, C.A.; Sinclair, J.H.; Alcami, A. The UL144 gene product of human cytomegalovirus activates NF-κB via a TRAF6-dependent mechanism. EMBO J. 2006, 25, 4390–4399. [Google Scholar] [CrossRef] [PubMed]
- Yoshie, O.; Imai, T.; Nomiyama, H. Chemokines in immunity. In Advances in Immunology; Elsevier: New York, NY, USA, 2001; Volume 78, pp. 57–110. [Google Scholar]
- Cha, T.-A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [PubMed]
- Gealy, C.; Humphreys, C.; Dickinson, V.; Stinski, M.; Caswell, R. An activation-defective mutant of the human cytomegalovirus IE2P86 protein inhibits NF-κB-mediated stimulation of the human interleukin-6 promoter. J. Gen. Virol. 2007, 88, 2435–2440. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Atkins, E.; Nakayama, T.; Yoshie, O.; Groves, I.; Alcami, A.; Sinclair, J. NF-κB-mediated activation of the chemokine CCL22 by the product of the human cytomegalovirus gene UL144 escapes regulation by viral IE86. J. Virol. 2008, 82, 4250–4256. [Google Scholar] [CrossRef] [PubMed]
- Nachtwey, J.; Spencer, J.V. Hcmv il-10 suppresses cytokine expression in monocytes through inhibition of NF-κB. Viral Immunol. 2008, 21, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Saccani, S.; Izotova, L.S.; Mirochnitchenko, O.V.; Pestka, S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvil-10). Proc. Natl. Acad. Sci. USA 2000, 97, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.C.; Logsdon, N.J.; Josephson, K.; Cook, J.; Barry, P.A.; Walter, M.R. Crystal structure of human cytomegalovirus IL-10 bound to soluble human IL-10R1. Proc. Natl. Acad. Sci. USA 2002, 99, 9404–9409. [Google Scholar] [CrossRef] [PubMed]
- Sester, M.; Sester, U.; Gärtner, B.; Kubuschok, B.; Girndt, M.; Meyerhans, A.; Köhler, H. Sustained high frequencies of specific CD4 T cells restricted to a single persistent virus. J. Virol. 2002, 76, 3748–3755. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.W.; Barry, P.A.; Szubin, R.; Wang, D.; Baumgarth, N. Human cytomegalovirus suppresses type I interferon secretion by plasmacytoid dendritic cells through its interleukin 10 homolog. Virology 2009, 390, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, J.V.; Cadaoas, J.; Castillo, P.R.; Saini, V.; Slobedman, B. Stimulation of b lymphocytes by cmvil-10 but not lacmvil-10. Virology 2008, 374, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Grey, F.; Antoniewicz, A.; Allen, E.; Saugstad, J.; McShea, A.; Carrington, J.C.; Nelson, J. Identification and characterization of human cytomegalovirus-encoded microRNAs. J. Virol. 2005, 79, 12095–12099. [Google Scholar] [CrossRef] [PubMed]
- Hook, L.; Hancock, M.; Landais, I.; Grabski, R.; Britt, W.; Nelson, J.A. Cytomegalovirus microRNAs. Curr. Opin. Virol. 2014, 7, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, T.J.; Arnold, J.D.; Spector, D.H.; Yeo, G.W. High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J. Virol. 2012, 86, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.H.; Hook, L.M.; Mitchell, J.; Nelson, J.A. Human cytomegalovirus microRNAs MIR-US5-1 and MIR-UL112-3p block proinflammatory cytokine production in response to NF-κB-activating factors through direct downregulation of IKKΑ and IKKΒ. mBio 2017, 8, e00109-00117. [Google Scholar] [CrossRef] [PubMed]
- Hook, L.M.; Grey, F.; Grabski, R.; Tirabassi, R.; Doyle, T.; Hancock, M.; Landais, I.; Jeng, S.; McWeeney, S.; Britt, W.; et al. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 2014, 15, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Lau, B.; Poole, E.; Krishna, B.; Montanuy, I.; Wills, M.R.; Murphy, E.; Sinclair, J. The expression of human cytomegalovirus microRNA mir-ul148d during latent infection in primary myeloid cells inhibits activin a-triggered secretion of IL-6. Sci. Rep. 2016, 6, 31205. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boomker, J.M.; The, T.H.; de Leij, L.F.M.H.; Harmsen, M.C. The human cytomegalovirus-encoded receptor US28 increases the activity of the major immediate-early promoter/enhancer. Virus Res. 2006, 118, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Beisser, P.S.; Laurent, L.; Virelizier, J.-L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [PubMed]
- Casarosa, P.; Bakker, R.A.; Verzijl, D.; Navis, M.; Timmerman, H.; Leurs, R.; Smit, M.J. Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J. Biol. Chem. 2001, 276, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. mBio 2017, 8, e01754-17. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Tan, S.Y.; Poole, E.; Talbot, S.; Antrobus, R.; Smith, D.L.; Montag, C.; Gygi, S.P.; Sinclair, J.H.; Lehner, P.J. Latency-associated degradation of the mrp1 drug transporter during latent human cytomegalovirus infection. Science 2013, 340, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Montag, C.; Wagner, J.A.; Gruska, I.; Vetter, B.; Wiebusch, L.; Hagemeier, C. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to TNFα signaling by upregulating TNFα receptor 1 cell surface expression. J. Virol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Le, V.T.K.; Trilling, M.; Hengel, H. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb’-encoded modulation of TNF-signaling. J. Virol. 2011, 85, 13260–13270. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Caviness, K.; Albright, E.R.; Lee, J.-H.; Gelbmann, C.B.; Rak, M.; Goodrum, F.; Kalejta, R.F. Long and short isoforms of the human cytomegalovirus UL138 protein silence ie1 transcription and promote latency. J. Virol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Albright, E.R.; Lee, J.-H.; Jacobs, D.; Kalejta, R.F. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci. Adv. 2015, 1, e1501164. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
HCMV-mediated modulation of Interferon signaling.

Figure 2.
HCMV-mediated modulation of NFκB signaling

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Goodwin, C.M.; Ciesla, J.H.; Munger, J. Who’s Driving? Human Cytomegalovirus, Interferon, and NFκB Signaling. Viruses 2018, 10, 447. https://0-doi-org.brum.beds.ac.uk/10.3390/v10090447
AMA Style
Goodwin CM, Ciesla JH, Munger J. Who’s Driving? Human Cytomegalovirus, Interferon, and NFκB Signaling. Viruses. 2018; 10(9):447. https://0-doi-org.brum.beds.ac.uk/10.3390/v10090447
Chicago/Turabian StyleGoodwin, Christopher M., Jessica H. Ciesla, and Joshua Munger. 2018. "Who’s Driving? Human Cytomegalovirus, Interferon, and NFκB Signaling" Viruses 10, no. 9: 447. https://0-doi-org.brum.beds.ac.uk/10.3390/v10090447
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.