High Fidelity Deep Sequencing Reveals No Effect of ATM, ATR, and DNA-PK Cellular DNA Damage Response Pathways on Adenovirus Mutation Rate

Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus and Cells
2.2. DDR Kinase Inhibition
2.3. Cytotoxicity
2.4. DDR Activation Measured by Immunofluorescence
2.5. Virus Titration
2.6. Serial Virus Transfers
2.7. Viral DNA Purification
2.8. Duplex Sequencing
2.9. Detection of Deletions
3. Results
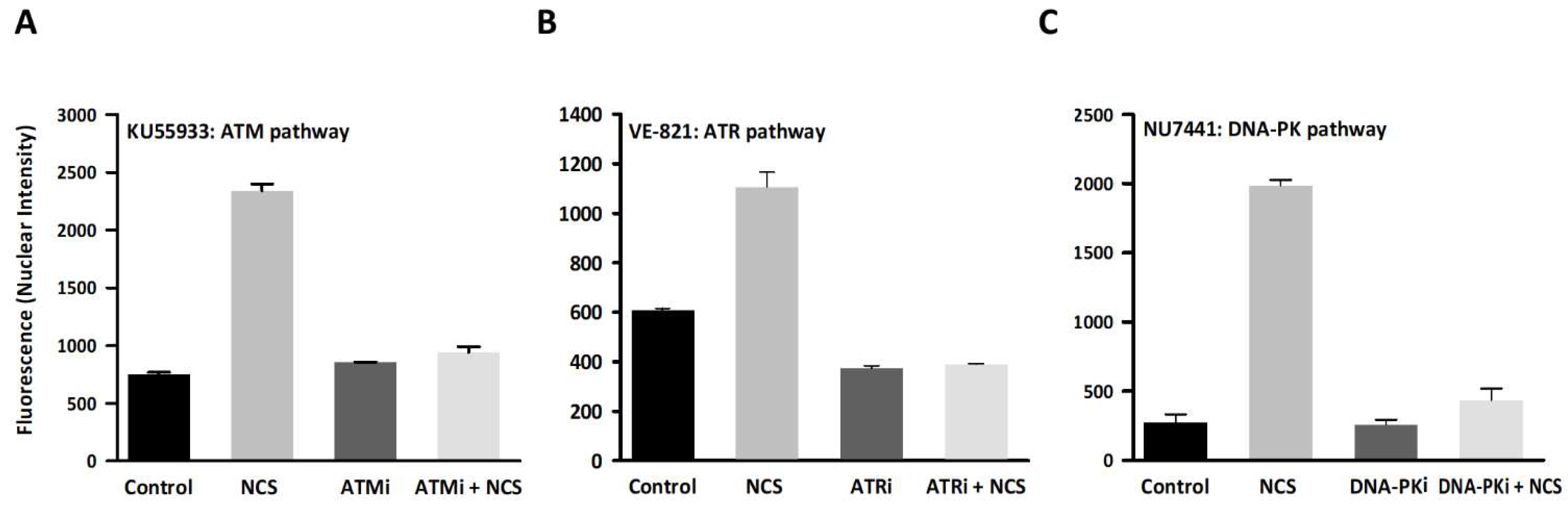
3.1. DDR Pathway Inhibition
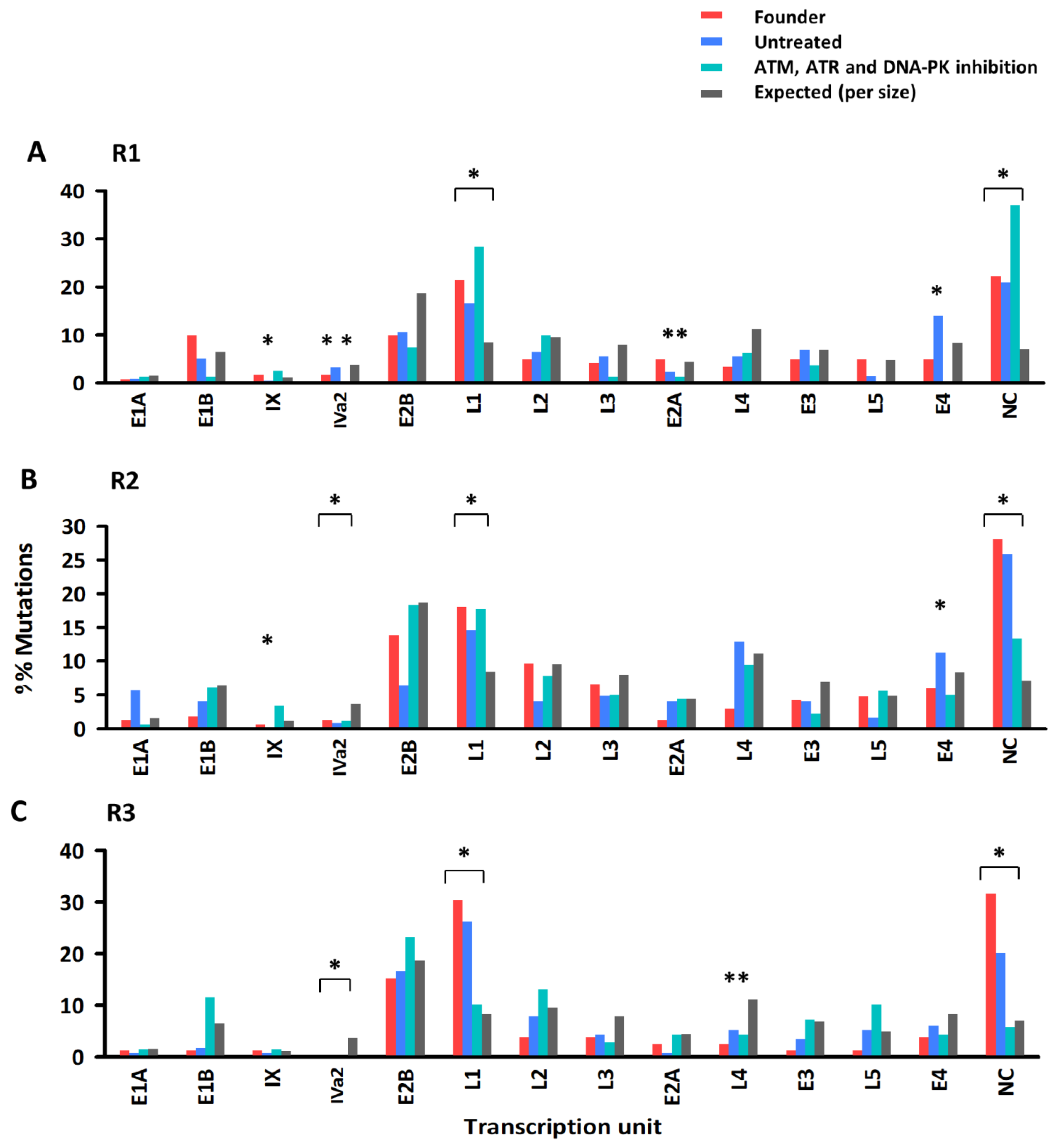
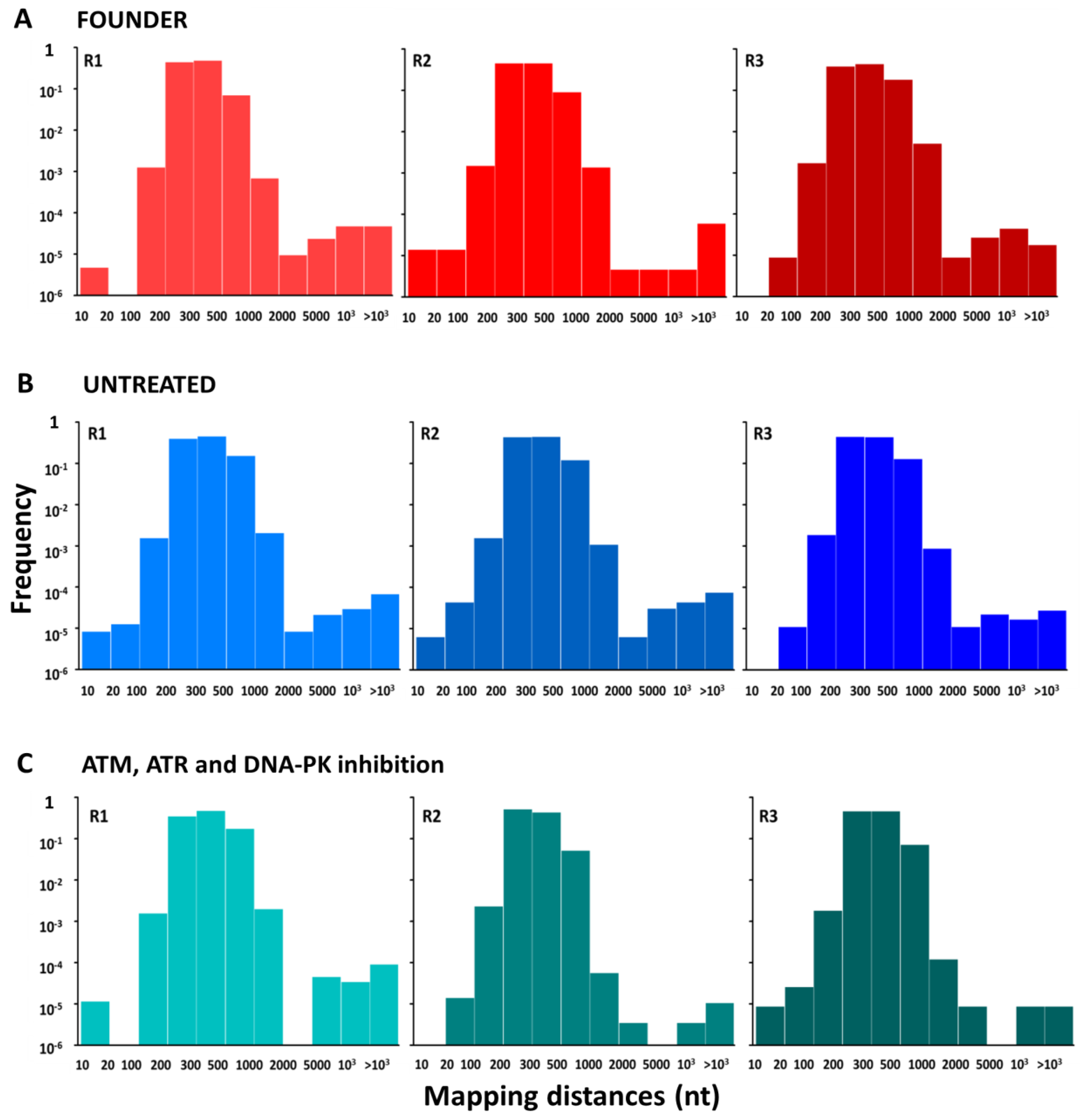
3.2. Effect of DDR Inhibition on the Accumulation of Genetic Diversity in Ad5
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and concepts in RNA virus population dynamics and evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Holmes, E.C. Evolutionary virology at 40. Genetics 2018, 210, 1151–1162. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Sanjuán, R. From Molecular Genetics to phylodynamics: Evolutionary relevance of mutation rates across viruses. PLoS Pathog. 2012, 8, e1002685. [Google Scholar] [CrossRef]
- Jiricny, J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef]
- Pereira-Gómez, M.; Sanjuán, R. Effect of mismatch repair on the mutation rate of bacteriophage ϕX174. Virus Evol. 2015, 1, vev010. [Google Scholar] [CrossRef]
- Trigg, B.J.; Ferguson, B.J. Functions of DNA damage machinery in the innate immune response to DNA virus infection. Curr. Opin. Virol. 2015, 15, 56–62. [Google Scholar] [CrossRef]
- Dybas, J.M.; Herrmann, C.; Weitzman, M.D. Ubiquitination at the interface of tumor viruses and DNA damage responses. Curr. Opin. Virol. 2018, 32, 40–47. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Luftig, M.A. Viruses and the DNA damage response: Activation and antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef]
- Chaurushiya, M.S.; Weitzman, M.D. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair 2009, 8, 1166–1176. [Google Scholar] [CrossRef] [Green Version]
- Lilley, C.E.; Schwartz, R.A.; Weitzman, M.D. Using or abusing: Viruses and the cellular DNA damage response. Trends Microbiol. 2007, 15, 119–126. [Google Scholar] [CrossRef]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, D.E.; Weller, S.K. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 2004, 78, 4783–4796. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Bloom, D.C.; Fisher, C. The ATM and Rad3-related (ATR) protein kinase pathway is activated by Herpes Simplex Virus 1 and required for efficient viral replication. J. Virol. 2017, 92, e01884-17. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.; You, J.; Benjamin, T.L. Induction and utilization of an ATM signaling pathway by polyomavirus. J. Virol. 2005, 79, 13007–13017. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dodson, G.E.; Shaikh, S.; Rundell, K.; Tibbetts, R.S. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem. 2005, 280, 40195–40200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Madden-Fuentes, R.J.; Lou, B.X.; Pipas, J.M.; Gerhardt, J.; Rigell, C.J.; Fanning, E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J. Virol. 2008, 82, 5316–5328. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA replication and the host DNA damage response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef]
- Liu, Y.; Shevchenko, A.; Shevchenko, A.; Berk, A.J. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 2005, 79, 14004–14016. [Google Scholar] [CrossRef]
- Araujo, F.D.; Stracker, T.H.; Carson, C.T.; Lee, D.V.; Weitzman, M.D. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic Aggresomes. J. Virol. 2005, 79, 11382–11391. [Google Scholar] [CrossRef]
- Baker, A.; Rohleder, K.J.; Hanakahi, L.A.; Ketner, G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 2007, 81, 7034–7040. [Google Scholar] [CrossRef]
- Orazio, N.I.; Naeger, C.M.; Karlseder, J.; Weitzman, M.D. The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J. Virol. 2011, 85, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Patel, R.N.; Forrester, N.A.; Theil, K.; Groitl, P.; Stewart, G.S.; Taylor, A.M.; Morgan, I.M.; Dobner, T.; Grand, R.J.; et al. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc. Natl. Acad. Sci. USA 2010, 107, 12251–12256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risso-Ballester, J.; Cuevas, J.M.; Sanjuán, R. Genome-wide estimation of the spontaneous mutation rate of human adenovirus 5 by high-fidelity deep sequencing. PLoS Pathog. 2016, 12, e1006013. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.C.; Risques, R.A.; et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc 2014, 9, 2586–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Valérie, B.; Herniou, E.A. Ultra deep sequencing of a baculovirus population reveals widespread genomic variations. Viruses 2015, 7, 3625–3646. [Google Scholar] [CrossRef] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 2012, 13, 1072–1081. [Google Scholar] [CrossRef] [Green Version]
- Leahy, J.J.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Richardson, C.; Rigoreau, L.; Smith, G.C. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett. 2004, 14, 6083–6087. [Google Scholar] [CrossRef]
- Beerman, T.A.; Mueller, G.; Grimmond, H. Effects of neocarzinostatin on chromatin in HeLa S3 nuclei. Mol. Pharmacol. 1983, 23, 493–499. [Google Scholar]
- Kappen, L.S.; Ellenberger, T.E.; Goldberg, I.H. Mechanism and base specificity of DNA Breakage in intact cells by neocarzinostatin. Biochemistry 1987, 26, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1988, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Lim, D. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Minn, K.; Chen, J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J. Biol. Chem. 2004, 279, 9677–9680. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef]
- Mukherjee, B.; Kessinger, C.; Kobayashi, J.; Chen, B.P.; Chen, D.J.; Chatterjee, A.; Burma, S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair 2006, 5, 575–590. [Google Scholar] [CrossRef]
- Reitsema, T.; Klokov, D.; Banáth, J.P.; Olive, P.L. DNA-PK is responsible for enhanced phosphorylation of histone H2AX under hypertonic conditions. DNA Repair 2005, 4, 1172–1181. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Wang, H.; Böcker, W.; Iliakis, G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell. Physiol. 2005, 202, 492–502. [Google Scholar] [CrossRef]
- King, A.J.; Teertstra, W.R.; van der Vliet, P.C. Dissociation of the protein primer and DNA polymerase after Initiation of adenovirus DNA replication. J. Biol. Chem. 1997, 272, 24617–24623. [Google Scholar] [CrossRef]
- Brenkman, A.B.; Breure, E.C.; van der Vliet, P.C. Molecular architecture of adenovirus DNA polymerase and location of the protein primer. J. Virol. 2002, 76, 8200–8207. [Google Scholar] [CrossRef]
- Liu, H.; Naismith, J.H.; Hay, R.T. Adenovirus DNA replication. Curr. Top. Microbiol. Immunol 2003, 272, 131–164. [Google Scholar]
- Uil, T.G.; Vellinga, J.; de Vrij, J.; van den Hengel, S.K.; Rabelink, M.J.; Cramer, S.J.; Eekels, J.J.; Ariyurek, Y.; van Galen, M.; Hoeben, R.C. Directed adenovirus evolution using engineered mutator viral polymerases. Nucleic Acids Res. 2011, 39, e30. [Google Scholar] [CrossRef]
- Cvijović, I.; Ba, A.A.N.; Desai, M.M. Experimental studies of evolutionary dynamics in microbes. Trends Genet. 2018, 34, 693–703. [Google Scholar] [CrossRef]
- Pancholi, N.J.; Price, A.M.; Weitzman, M.D. Take your PIKK: Tumour viruses and DNA damage response pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160269. [Google Scholar] [CrossRef]
- Malenovska, H. Virus quantitation by transmission electron microscopy, TCID50, and the role of timing virus harvesting: A case study of three animal viruses. J. Virol Methods 2013, 191, 136–140. [Google Scholar] [CrossRef]
- Reid, G.G.; Milne, E.W.; Coggins, L.W.; Wilson, N.J.; Smith, K.T.; Shepherd, A.J. Comparison of electron microscopic techniques for enumeration of endogenous retrovirus in mouse and Chinese hamster cell lines used for production of biologics. J. Virol Methods 2003, 108, 91–96. [Google Scholar] [CrossRef]
- Kramberger, P.M.; Ciringer, A.S.; Peterka, M. Evaluation of nanoparticle tracking analysis for total virus particle determination. Virol J. 2012, 9, 265. [Google Scholar] [CrossRef]
- Nikitin, N.; Trifonova, E.; Evtushenko, E.; Kirpichnikov, M.; Atabekov, J.; Karpova, O. Comparative Study of Non-Enveloped Icosahedral Viruses Size. PLoS ONE 2015, 10, e0142415. [Google Scholar] [CrossRef]
- Blundell, E.L.C.J.; Mayne, L.J.; Billinge, E.R.; Platt, M. Emergence of tunable resistive pulse sensing as a biosensor. Anal. Methods 2015, 7, 7055–7066. [Google Scholar] [CrossRef] [Green Version]
- William, S.M.W.; Ann, T.E. Adenovirus Methods and Protocols, 2nd ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 185–192. [Google Scholar]
- Wold, W.S.M.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Ahi, Y.S.; Bangari, D.S.; Mittal, S.K. Adenoviral vector immunity: Its implications and circumvention strategies. Curr. Gene Ther. 2011, 11, 307–320. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Experiment | 1 | 2 | 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Virus population | Founder | Untreated | DDRi 1 | Founder | Untreated | DDRi 1 | Founder | Untreated | DDRi 1 |
| Polymorphic sites | 122 | 215 | 81 | 171 | 125 | 182 | 80 | 113 | 68 |
| Total mutation count | 159 | 396 | 118 | 272 | 243 | 320 | 96 | 408 | 92 |
| Mbp sequenced | 42.9 | 43.6 | 23.6 | 49.8 | 33.2 | 48.9 | 18.8 | 78.1 | 28.5 |
| Mutation frequency (×106) | 2.8 | 4.9 | 3.4 | 3.4 | 3.8 | 3.7 | 4.3 | 1.4 | 2.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Risso-Ballester, J.; Sanjuán, R. High Fidelity Deep Sequencing Reveals No Effect of ATM, ATR, and DNA-PK Cellular DNA Damage Response Pathways on Adenovirus Mutation Rate. Viruses 2019, 11, 938. https://0-doi-org.brum.beds.ac.uk/10.3390/v11100938
Risso-Ballester J, Sanjuán R. High Fidelity Deep Sequencing Reveals No Effect of ATM, ATR, and DNA-PK Cellular DNA Damage Response Pathways on Adenovirus Mutation Rate. Viruses. 2019; 11(10):938. https://0-doi-org.brum.beds.ac.uk/10.3390/v11100938
Chicago/Turabian StyleRisso-Ballester, Jennifer, and Rafael Sanjuán. 2019. "High Fidelity Deep Sequencing Reveals No Effect of ATM, ATR, and DNA-PK Cellular DNA Damage Response Pathways on Adenovirus Mutation Rate" Viruses 11, no. 10: 938. https://0-doi-org.brum.beds.ac.uk/10.3390/v11100938