Isolation and Characterization of Clinical RSV Isolates in Belgium during the Winters of 2016–2018

 , , ,
, , ,  , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Cells and Viruses
2.2. Virus Isolation from Clinical Samples
2.3. RSV-A and RSV-B Subtyping
2.4. Gene Nucleotide Sequencing and Phylogenetic Analysis
2.5. Viral Replication Kinetics
2.6. Infectious Virus Production
2.7. Thermal Stability Assay
2.8. Cell-to-Cell Fusion Assay
2.9. Plaque Reduction Assay
2.10. Mucin mRNA Expression Assay
2.11. Fluorescence Microscopy and Image Analysis
2.12. Statistical Analysis
3. Results
3.1. Clinical Samples and Detection of RSV
3.2. Phylogenetic Analysis
3.3. Viral Replication Kinetics
3.4. Thermal Stability
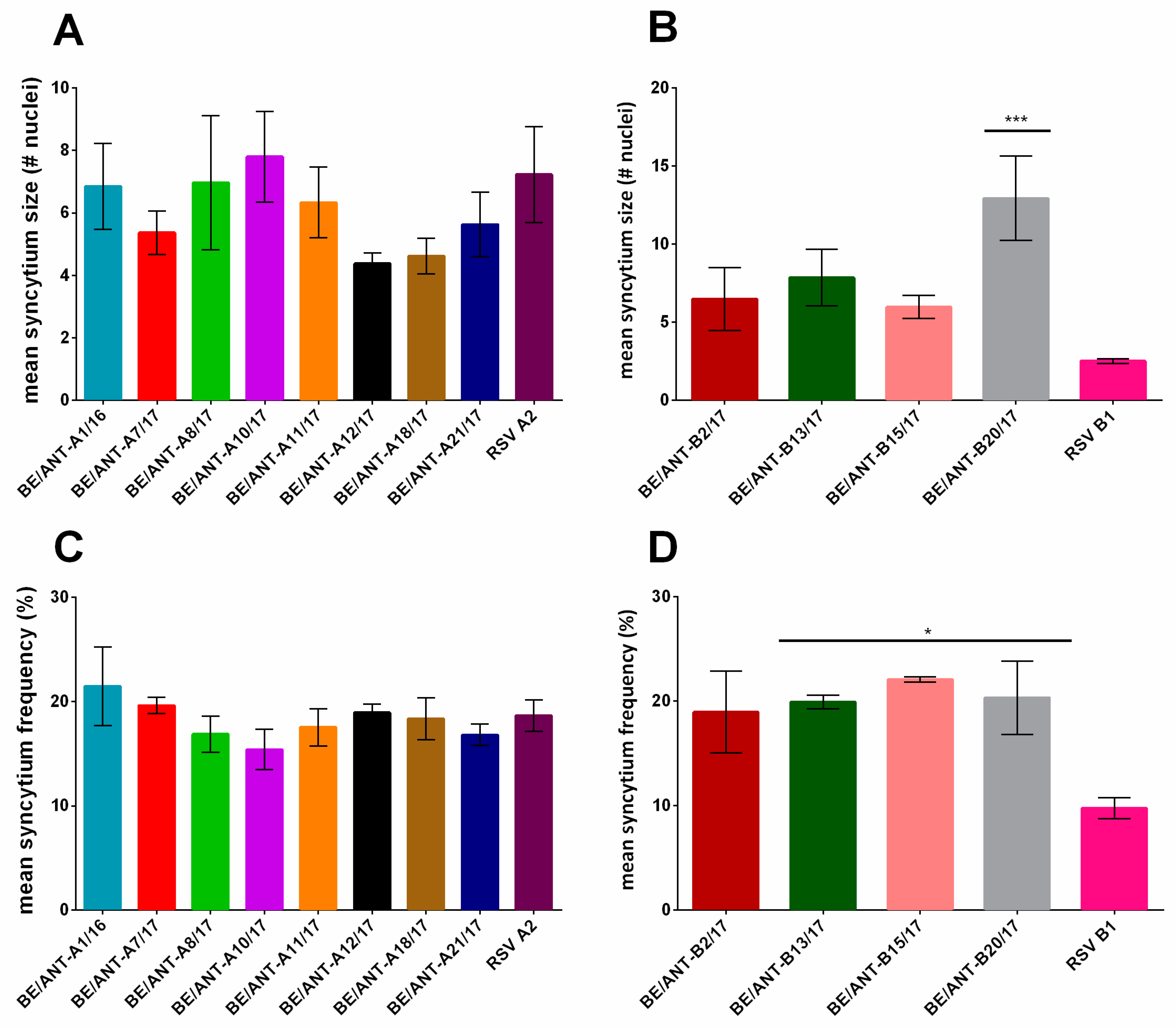
3.5. Cell to Cell Fusion
3.6. Plaque Reduction by Palivizumab
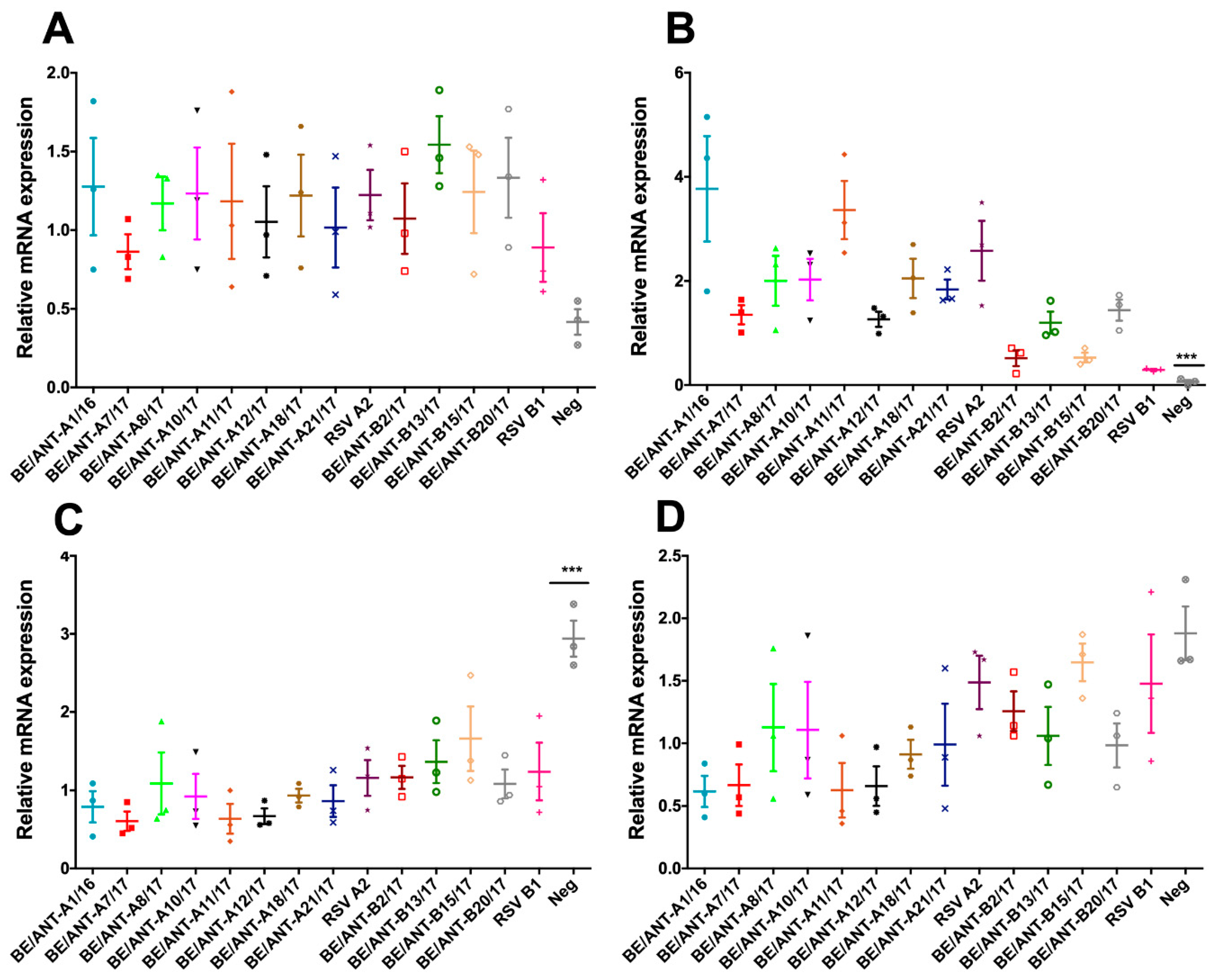
3.7. Mucin Expression
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Collins, P.L.; Graham, B.S. Viral and Host Factors in Human Respiratory Syncytial Virus Pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef] [Green Version]
- Borchers, A.T.; Chang, C.; Gershwin, M.E.; Gershwin, L.J. Respiratory Syncytial Virus—A Comprehensive Review. Clin. Rev. Allergy Immunol. 2013, 45, 331–379. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.F.; Lam, D.S.Y.; Miu, T.Y.; Hon, K.L.; Chau, C.S.K.; Ku, S.W.; Lee, R.S.Y.; Chow, P.Y.; Chiu, W.K.; Ng, D.K.K. Epidemiology and risk factors for severe respiratory syncytial virus infections requiring pediatric intensive care admission in Hong Kong children. Infection 2014, 42, 343–350. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory Syncytial Virus Infection in Elderly and High-Risk Adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Ebbert, J.O.; Limper, A.H. Respiratory Syncytial Virus Pneumonitis in Immunocompromised Adults: Clinical Features and Outcome. Respiration 2005, 72, 263–269. [Google Scholar] [CrossRef]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The Burden of Respiratory Syncytial Virus Infection in Young Children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Johnson, S.; Oliver, C.; Prince, G.A.; Hemming, V.G.; Pfarr, D.S.; Wang, S.C.; Dormitzer, M.; O’Grady, J.; Koenig, S.; Tamura, J.K.; et al. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 1997, 176, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S. Neutralizing epitopes on the respiratory syncytial virus fusion glycoprotein. Curr. Opin. Virol. 2015, 11, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Feldman, S.A.; Hendry, R.M.; Beeler, J.A. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J. Virol. 1999, 73, 6610–6617. [Google Scholar]
- Feldman, S.A.; Audet, S.; Beeler, J.A. The Fusion Glycoprotein of Human Respiratory Syncytial Virus Facilitates Virus Attachment and Infectivity via an Interaction with Cellular Heparan Sulfate. J. Virol. 2000, 74, 6442–6647. [Google Scholar] [CrossRef]
- Johnson, P.R.; Spriggs, M.K.; Olmsted, R.A.; Collins, P.L. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: Extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 5625–5629. [Google Scholar] [CrossRef] [PubMed]
- Peret, T.C.T.; Golub, J.A.; Anderson, L.J.; Hall, C.B.; Schnabel, K.C. Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J. Gen. Virol. 1998, 79, 2221–2229. [Google Scholar] [CrossRef]
- Peret, T.C.T.; Hall, C.B.; Hammond, G.W.; Piedra, P.A.; Storch, G.A.; Sullender, W.M.; Tsou, C.; Anderson, L.J. Circulation Patterns of Group A and B Human Respiratory Syncytial Virus Genotypes in 5 Communities in North America. J. Infect. Dis. 2000, 181, 1891–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, M.; Madhi, S.A.; Tiemessen, C.T.; Schoub, B.D. Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: Identification of new subgroup A and B genotypes. J. Gen. Virol. 2001, 82, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Eshaghi, A.; Duvvuri, V.R.; Lai, R.; Nadarajah, J.T.; Li, A.; Patel, S.N.; Low, D.E.; Gubbay, J.B. Genetic Variability of Human Respiratory Syncytial Virus A Strains Circulating in Ontario: A Novel Genotype with a 72 Nucleotide G Gene Duplication. PLoS ONE 2012, 7, e32807. [Google Scholar] [CrossRef]
- Pretorius, M.A.; Van Niekerk, S.; Tempia, S.; Moyes, J.; Cohen, C.; Madhi, S.A.; Venter, M.; SARI Surveillance Group. Replacement and Positive Evolution of Subtype A and B Respiratory Syncytial Virus G-Protein Genotypes From 1997–2012 in South Africa. J. Infect. Dis 2013, 208, S227–S237. [Google Scholar] [CrossRef]
- Ren, L.; Xia, Q.; Xiao, Q.; Zhou, L.; Zang, N.; Long, X.; Xie, X.; Deng, Y.; Wang, L.; Fu, Z.; et al. The genetic variability of glycoproteins among respiratory syncytial virus subtype A in China between 2009 and 2013. Infect. Genet. Evol. 2014, 27, 339–347. [Google Scholar] [CrossRef]
- Venter, M.; Collinson, M.; Schoub, B.D. Molecular epidemiological analysis of community circulating respiratory syncytial virus in rural South Africa: Comparison of viruses and genotypes responsible for different disease manifestations. J. Med. Virol. 2002, 68, 452–461. [Google Scholar] [CrossRef]
- Dapat, I.C.; Shobugawa, Y.; Sano, Y.; Saito, R.; Sasaki, A.; Suzuki, Y.; Kumaki, A.; Zaraket, H.; Dapat, C.; Oguma, T.; et al. New Genotypes within Respiratory Syncytial Virus Group B Genotype BA in Niigata, Japan. J. Clin. Microbiol. 2010, 48, 3423–3427. [Google Scholar] [CrossRef] [Green Version]
- Diallo, M.S.; Fromer, N.A.; Jhon, M.S. Nanotechnology for sustainable development: Retrospective and outlook. J. Nanoparticle Res. 2013, 15, 2044. [Google Scholar] [CrossRef]
- Auksornkitti, V.; Kamprasert, N.; Thongkomplew, S.; Suwannakarn, K.; Theamboonlers, A.; Samransamruajkij, R.; Poovorawan, Y. Molecular characterization of human respiratory syncytial virus, 2010-2011: Identification of genotype ON1 and a new subgroup B genotype in Thailand. Arch. Virol. 2014, 159, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Xiao, Q.; Zhou, L.; Xia, Q.; Liu, E. Molecular characterization of human respiratory syncytial virus subtype B: A novel genotype of subtype B circulating in China. J. Med. Virol. 2015, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Baek, Y.H.; Choi, E.H.; Song, M.-S.; Pascua, P.N.Q.; Kwon, H.; Park, S.-J.; Lee, J.H.; Woo, S.I.; Ahn, B.H.; Han, H.S.; et al. Prevalence and genetic characterization of respiratory syncytial virus (RSV) in hospitalized children in Korea. Arch. Virol. 2012, 157, 1039–1050. [Google Scholar] [CrossRef]
- Khor, C.-S.; Sam, I.-C.; Hooi, P.-S.; Chan, Y.-F. Displacement of predominant respiratory syncytial virus genotypes in Malaysia between 1989 and 2011. Infect. Genet. Evol. 2013, 14, 357–360. [Google Scholar] [CrossRef]
- Collins, P.L.; Karron, R.A. Respiratory Syncytial Virus and Metapneumovirus. Fields Virol. 2013, 1, 1086. [Google Scholar]
- Gagliardi, T.B.; Criado, M.F.; Proença-Módena, J.L.; Saranzo, A.M.; Iwamoto, M.A.; de Paula, F.E.; Cardoso, R.S.; Delcaro, L.S.; Silva, M.L.; Câmara, A.A.; et al. Syncytia Induction by Clinical Isolates of Human Respiratory Syncytial Virus, A. Intervirology 2017, 60, 56–60. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway Mucus Function and Dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [Green Version]
- Ridley, C.; Thornton, D.J. Mucins: The frontline defence of the lung. Biochem. Soc. Trans. 2018, 46, 1099–1106. [Google Scholar]
- Morris, J.A.; Blount, R.E.; Savage, R.E. Recovery of Cytopathogenic Agent from Chimpanzees with Goryza. Exp. Biol. Med. 1956, 92, 544–549. [Google Scholar] [CrossRef]
- Chanock, R.; Roizman, B.; Myers, R. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). I. Isolation, properties and characterization. Am. J. Hyg. 1957, 66, 281–290. [Google Scholar] [PubMed]
- Van der Gucht, W.; Leemans, A.; De Schryver, M.; Heykers, A.; Caljon, G.; Maes, L.; Cos, P.; Delputte, P.L. Respiratory syncytial virus (RSV) entry is inhibited by serine protease inhibitor AEBSF when present during an early stage of infection. Virol. J. 2017, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Schepens, B.; Sedeyn, K.; Vande Ginste, L.; De Baets, S.; Schotsaert, M.; Roose, K.; Houspie, L.; Van Ranst, M.; Gilbert, B.; van Rooijen, N.; et al. Protection and mechanism of action of a novel human respiratory syncytial virus vaccine candidate based on the extracellular domain of small hydrophobic protein. EMBO Mol. Med. 2014, 6, 1436–1454. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.; Barbezange, C.; Van Gucht, S.; Weyckmans, J.; Fdillate, I.; Van Eycken, R.; Hamouda, A.; Bossuyt, N.; Quoilin, S.; Van Casteren, V.; et al. Virological Surveillance of Influenza in Belgium Season 2017-2018. Sciensano 2018, 40. [Google Scholar]
- Houspie, L.; Lemey, P.; Keyaerts, E.; Reijmen, E.; Vergote, V.; Vankeerberghen, A.; Vaeyens, F.; De Beenhouwer, H.; Van Ranst, M. Circulation of HRSV in Belgium: From Multiple Genotype Circulation to Prolonged Circulation of Predominant Genotypes. PLoS ONE 2013, 8, e60416. [Google Scholar] [CrossRef] [PubMed]
- Leemans, A.; Boeren, M.; Van der Gucht, W.; Pintelon, I.; Roose, K.; Schepens, B.; Saelens, X.; Bailey, D.; Martinet, W.; Caljon, G.; et al. Removal of the N-Glycosylation Sequon at Position N116 Located in p27 of the Respiratory Syncytial Virus Fusion Protein Elicits Enhanced Antibody Responses after DNA Immunization. Viruses 2018, 10, 426. [Google Scholar] [CrossRef]
- Stobart, C.C.; Rostad, C.A.; Ke, Z.; Dillard, R.S.; Hampton, C.M.; Strauss, J.D.; Yi, H.; Hotard, A.L.; Meng, J.; Pickles, R.J.; et al. A live RSV vaccine with engineered thermostability is immunogenic in cotton rats despite high attenuation. Nat. Commun. 2016, 7, 13916. [Google Scholar] [CrossRef]
- Baños-Lara, M.D.R.; Piao, B.; Guerrero-Plata, A. Differential Mucin Expression by Respiratory Syncytial Virus and Human Metapneumovirus Infection in Human Epithelial Cells. Mediators Inflamm. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Karron, R.A.; Buonagurio, D.A.; Georgiu, A.F.; Whitehead, S.S.; Adamus, J.E.; Clements-Mann, M.L.; Harris, D.O.; Randolph, V.B.; Udem, S.A.; Murphy, B.R.; et al. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: Clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. USA 1997, 94, 13961–13966. [Google Scholar] [CrossRef]
- Zimmer, G.; Trotz, I.; Herrler, G. N-Glycans of F Protein Differentially Affect Fusion Activity of Human Respiratory Syncytial Virus. J. Virol. 2001, 75, 4744–4751. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAME: | SUBTYPE: | GROWN ON: | VIRUS ISOLATED FROM: | EXPERIMENTS PERFORMED WITH: |
|---|---|---|---|---|
| BE/ANT-A1/16 | RSV-A | HEp-2 | Aspirate | Aspirate |
| BE/ANT-B2/17 | RSV-B | HEp-2 | Aspirate/Swab | Aspirate |
| BE/ANT-A7/17 | RSV-A | HEp-2 | Aspirate/Swab | Aspirate |
| BE/ANT-A8/17 | RSV-A | HEp-2 | Aspirate | Aspirate |
| BE/ANT-A10/17 | RSV-A | HEp-2 | Swab | Swab |
| BE/ANT-A11/17 | RSV-A | HEp-2 | Aspirate | Aspirate |
| BE/ANT-A12/17 | RSV-A | HEp-2 | Aspirate/Swab | Aspirate |
| BE/ANT-B13/17 | RSV-B | HEp-2 | Aspirate | Aspirate |
| BE/ANT-B15/17 | RSV-B | HEp-2 | Aspirate/Swab | Aspirate |
| BE/ANT-A18/17 | RSV-A | HEp-2 | Aspirate | Aspirate |
| BE/ANT-B20/17 | RSV-B | Vero | Aspirate | Aspirate |
| BE/ANT-A21/17 | RSV-A | HEp-2 | Aspirate/Swab | Aspirate |
| RSV A2 | RSV-A | HEp-2 | / | |
| RSV B1 | RSV-B | Vero | / |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Gucht, W.; Stobbelaar, K.; Govaerts, M.; Mangodt, T.; Barbezange, C.; Leemans, A.; De Winter, B.; Van Gucht, S.; Caljon, G.; Maes, L.; et al. Isolation and Characterization of Clinical RSV Isolates in Belgium during the Winters of 2016–2018. Viruses 2019, 11, 1031. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111031
Van der Gucht W, Stobbelaar K, Govaerts M, Mangodt T, Barbezange C, Leemans A, De Winter B, Van Gucht S, Caljon G, Maes L, et al. Isolation and Characterization of Clinical RSV Isolates in Belgium during the Winters of 2016–2018. Viruses. 2019; 11(11):1031. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111031
Chicago/Turabian StyleVan der Gucht, Winke, Kim Stobbelaar, Matthias Govaerts, Thomas Mangodt, Cyril Barbezange, Annelies Leemans, Benedicte De Winter, Steven Van Gucht, Guy Caljon, Louis Maes, and et al. 2019. "Isolation and Characterization of Clinical RSV Isolates in Belgium during the Winters of 2016–2018" Viruses 11, no. 11: 1031. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111031