A Multi-Omics Study of Chicken Infected by Nephropathogenic Infectious Bronchitis Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Strains
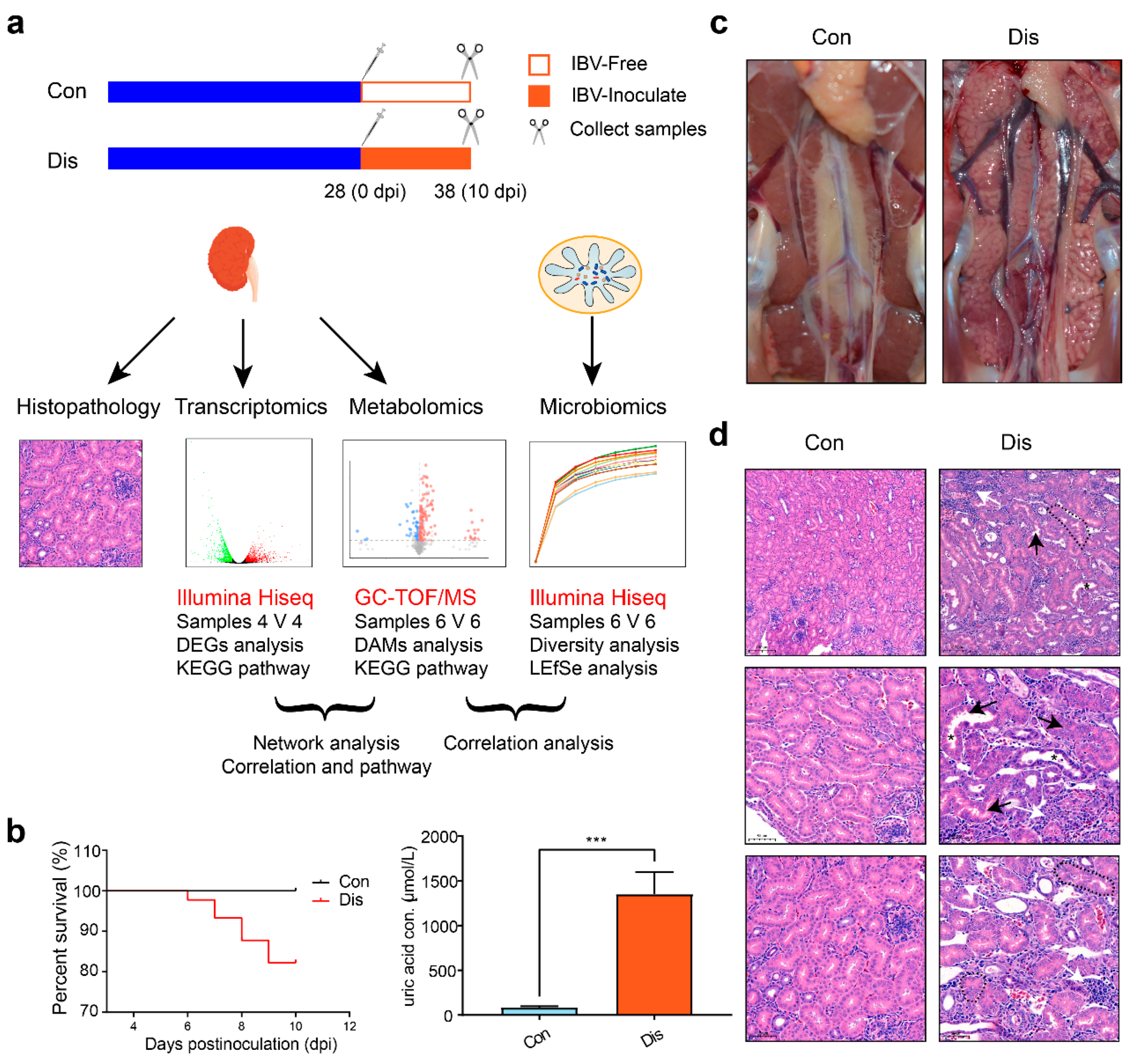
2.2. Experimental Design
2.3. Histopathology
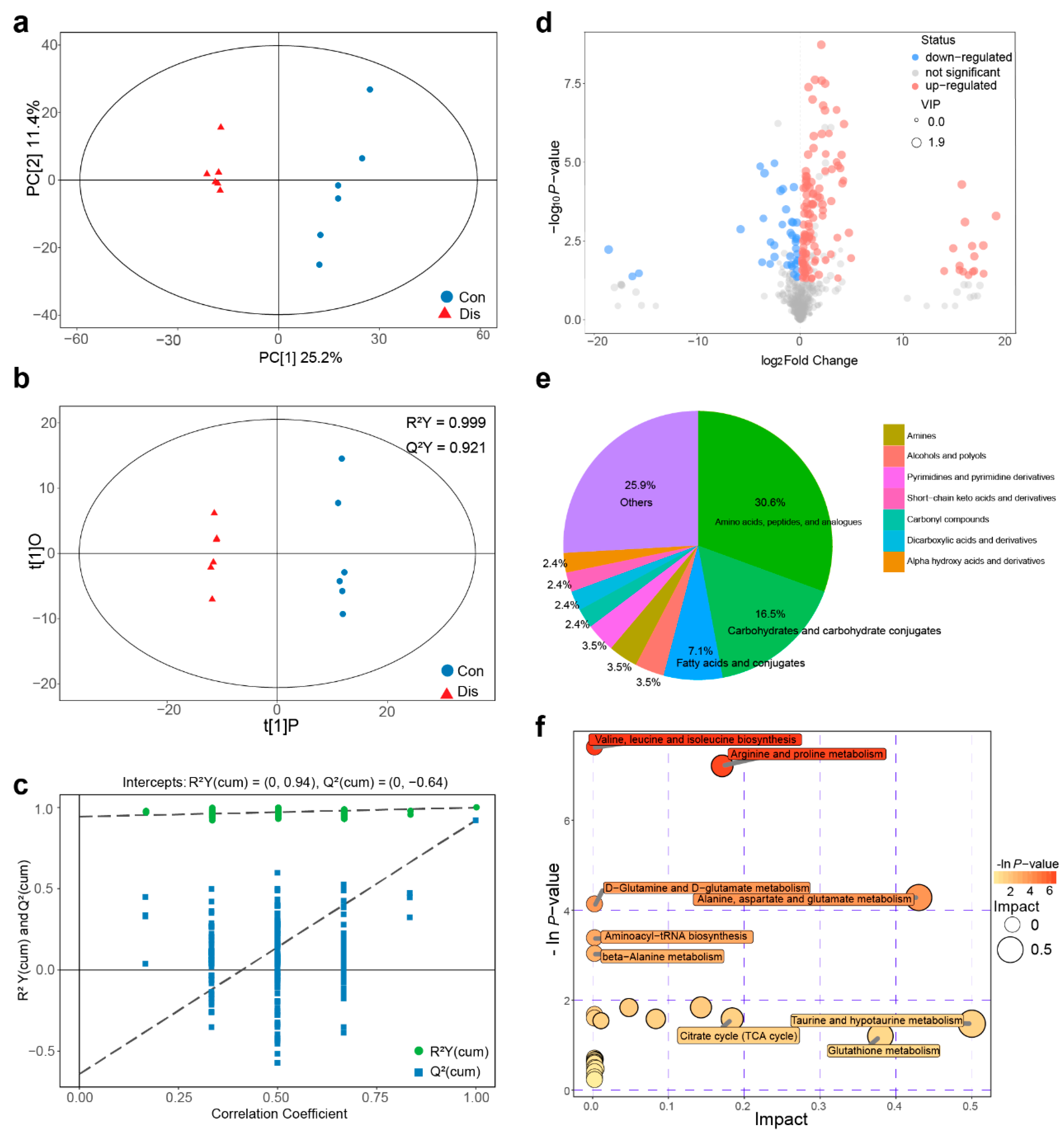
2.4. Metabolomics Analysis
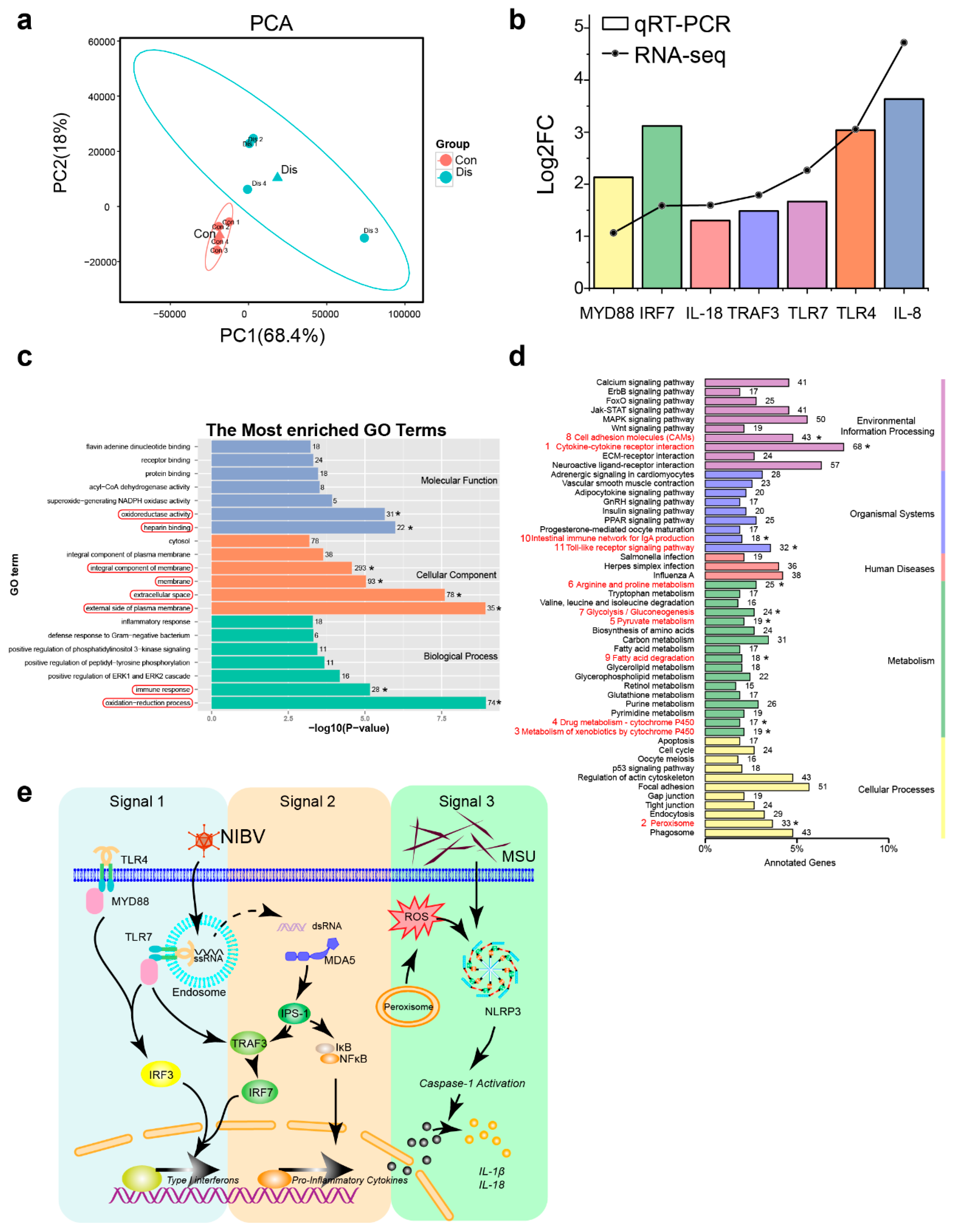
2.5. Transcriptomics Analysis
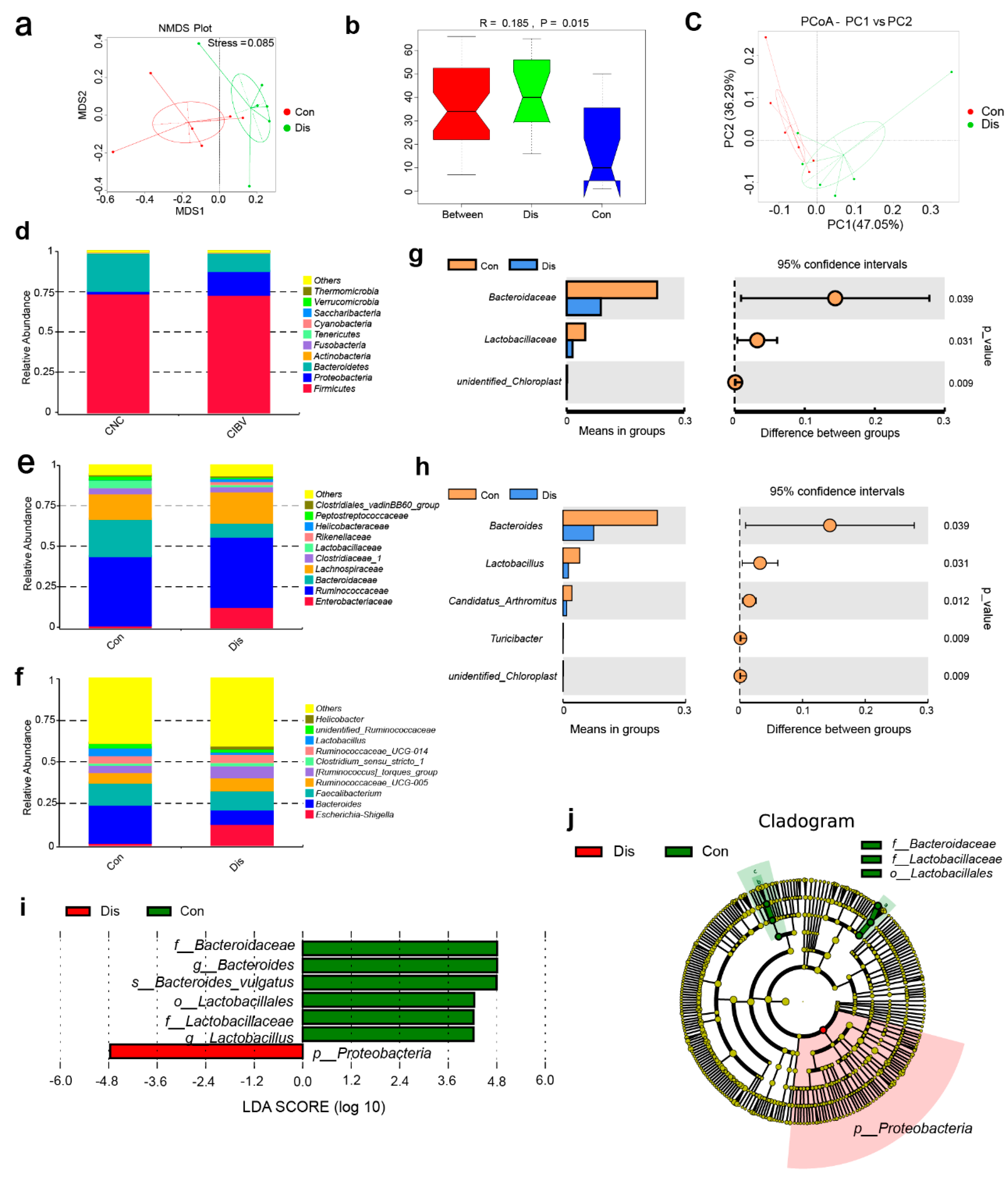
2.6. Microbiomics Analysis
2.7. Ethics Approval and Consent to Participate
3. Results
3.1. Clinical Signs and Pathology
3.2. NIBV Infection Altered Metabolic Profiling in the Kidney of Chickens
3.3. NIBV Infection Altered Transcription Profiling in the Kidney of Chickens
3.4. NIBV Infection Resulted in Gut Microbiota Dysbiosis
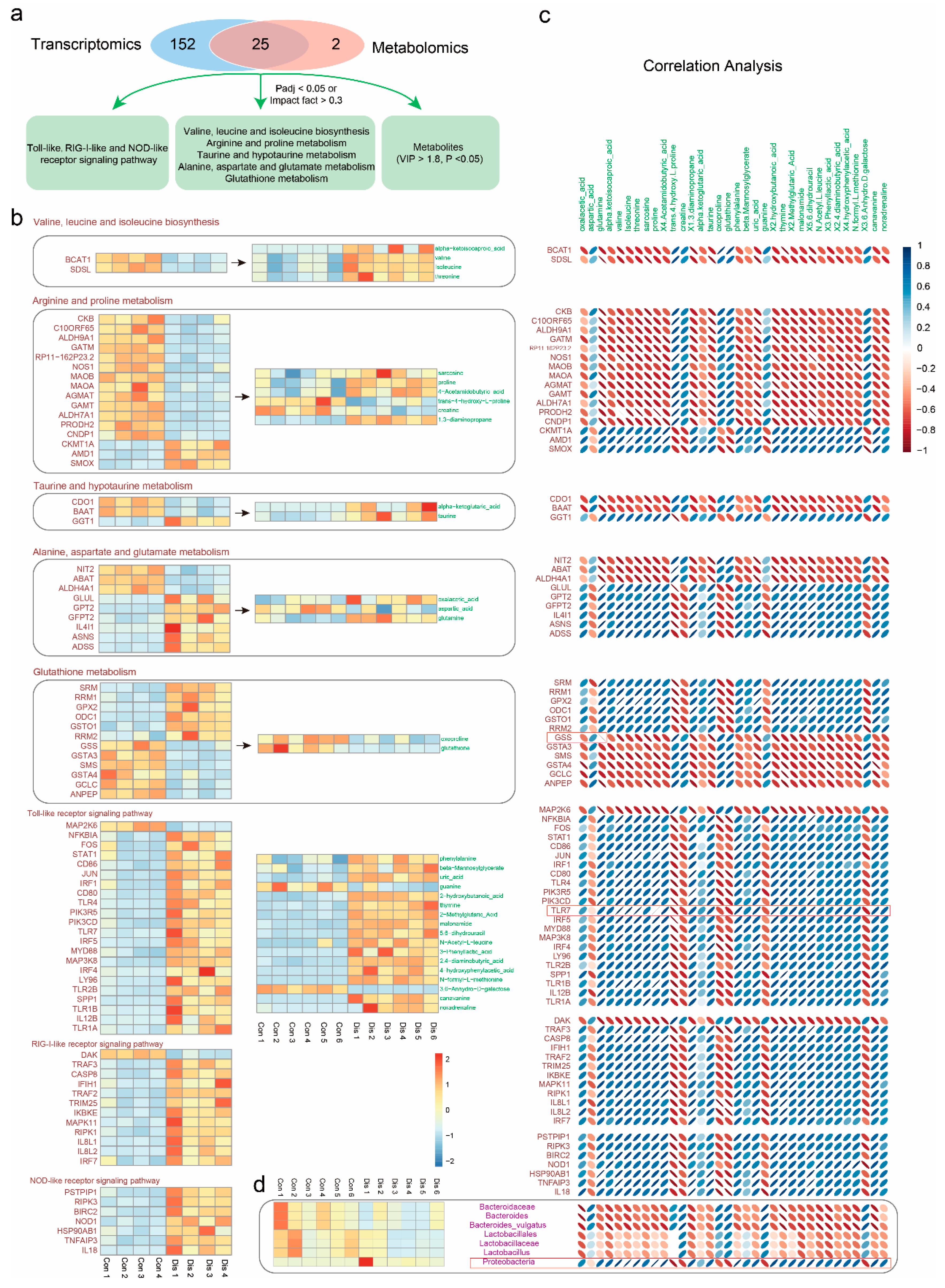
3.5. Correlation Analysis of Metabolomics and Transcriptomics Data
3.6. Correlation Analysis of Metabolomics and Microbiomics Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef]
- Roddy, E.; Choi, H.K. Epidemiology of gout. Rheum. Dis. Clin. N. Am. 2014, 40, 155–175. [Google Scholar] [CrossRef]
- Ejaz, S.; Kim, B.S.; Lim, C.W. Gout Induced by Intoxication of Sodium Bicarbonate in Korean Native Broilers. Drug Chem. Toxicol. 2005, 28, 245–261. [Google Scholar] [CrossRef]
- Guo, X.; Huang, K.; Tang, J. Clinicopathology of gout in growing layers induced by high calcium and high protein diets. Br. Poult. Sci. 2005, 46, 641–646. [Google Scholar] [CrossRef]
- Ziegler, A.F.; Ladman, B.S.; Dunn, P.A.; Schneider, A.; Davison, S.; Miller, P.G.; Lu, H.; Weinstock, D.; Salem, M.; Eckroade, R.J.; et al. Nephropathogenic infectious bronchitis in Pennsylvania chickens 1997–2000. Avian Dis. 2002, 46, 847–858. [Google Scholar] [CrossRef]
- Bulbule, N.R.; Mandakhalikar, K.D.; Kapgate, S.S.; Deshmukh, V.V.; Schat, K.A.; Chawak, M.M. Role of chicken astrovirus as a causative agent of gout in commercial broilers in India. Avian Pathol. 2013, 42, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.R.; Theuns, S.; Roukaerts, I.D.; Zeller, M.; Matthijnssens, J.; Nauwynck, H.J. Genetic Characterization of the Belgian Nephropathogenic Infectious Bronchitis Virus (NIBV) Reference Strain B1648. Viruses 2015, 7, 4488–4506. [Google Scholar] [CrossRef] [PubMed]
- Bayry, J.; Goudar, M.S.; Nighot, P.K.; Kshirsagar, S.G.; Ladman, B.S.; Gelb, J., Jr.; Ghalsasi, G.R.; Kolte, G.N. Emergence of a nephropathogenic avian infectious bronchitis virus with a novel genotype in India. J. Clin. Microbiol. 2005, 43, 916–918. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, Z.H.; Sleman, R.R.; Uthman, A.U. Isolation and molecular characterization of Sul/01/09 avian infectious bronchitis virus, indicates the emergence of a new genotype in the Middle East. Vet. Microbiol. 2011, 150, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zheng, Y.; Yang, Y.; Liu, C.; Geng, Q.; Luo, C.; Zhang, W.; Li, F. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 2018, 14, e1007009. [Google Scholar] [CrossRef]
- Wu, X.; Zhai, X.; Lai, Y.; Zuo, L.; Zhang, Y.; Mei, X.; Xiang, R.; Kang, Z.; Zhou, L.; Wang, H. Construction and Immunogenicity of Novel Chimeric Virus-Like Particles Bearing Antigens of Infectious Bronchitis Virus and Newcastle Disease Virus. Viruses 2019, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K.; Jackwood, M.; Jones, R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012, 41, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Naeem, K.; Hameed, A. Detection and seroprevalence of infectious bronchitis virus strains in commercial poultry in Pakistan. Poult. Sci. 2007, 86, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Nakayama, J.; Moriya, K.; Kawaratani, H.; Momoda, R.; Ito, K.; Iio, E.; Nojiri, S.; Fujiwara, K.; Yoneda, M.; et al. Gut Dysbiosis Associated With Hepatitis C Virus Infection. Clin. Infect. Dis. 2018, 67, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Oakley, B.B.; Lillehoj, H.S.; Kogut, M.H.; Kim, W.K.; Maurer, J.J.; Pedroso, A.; Lee, M.D.; Collett, S.R.; Johnson, T.J.; Cox, N.A. The chicken gastrointestinal microbiome. FEMS Microbiol. Lett. 2014, 360, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, M.J.; Constantinidou, C.; Cogan, T.A.; Bedford, M.R.; Penn, C.W.; Pallen, M.J. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS ONE 2014, 9, e91941. [Google Scholar] [CrossRef]
- Mishra, A.K.; Kumar, A.; Mishra, D.; Nath, V.S.; Jakse, J.; Kocabek, T.; Killi, U.K.; Morina, F.; Matousek, J. Genome-Wide Transcriptomic Analysis Reveals Insights into the Response to Citrus bark cracking viroid (CBCVd) in Hop (Humulus lupulus L.). Viruses 2018, 10, 570. [Google Scholar] [CrossRef]
- Zhang, T.; Feng, W.; Ye, J.; Li, Z.; Zhou, G. Metabolomic Changes in Sogatella furcifera under Southern rice black-streaked dwarf virus Infection and Temperature Stress. Viruses 2018, 10, 344. [Google Scholar] [CrossRef]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef]
- Sancesario, G.M.; Bernardini, S. Alzheimer’s disease in the omics era. Clin. Biochem. 2018, 59, 9–16. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2016, 2, 16180. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Sun, L.; Zhang, X. Integration of microbiome and epigenome to decipher the pathogenesis of autoimmune diseases. J. Autoimmun. 2017, 83, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Alexandar, V.; Nayar, P.G.; Murugesan, R.; Mary, B.; Darshana, P.; Shiek, S.S.J.A. CardioGenBase: A Literature Based Multi-Omics Database for Major Cardiovascular Diseases. PLoS ONE 2015, 10, e0143188. [Google Scholar] [CrossRef]
- Lin, H.; Huang, Q.; Guo, X.; Liu, P.; Liu, W.; Zou, Y.; Zhu, S.; Deng, G.; Kuang, J.; Zhang, C.; et al. Elevated level of renal xanthine oxidase mRNA transcription after nephropathogenic infectious bronchitis virus infection in growing layers. J. Vet. Sci. 2015, 16, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hao, R.; Du, X.; Deng, Y.; Sun, R.; Wang, Q. Metabolomics Responses of Pearl Oysters (Pinctada fucata martensii) Fed a Formulated Diet Indoors and Cultured With Natural Diet Outdoors. Front. Physiol. 2018, 9, 944. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Cao, H.; Xiao, Q.; Guo, X.; Zhuang, Y.; Zhang, C.; Wang, T.; Lin, H.; Song, Y.; Hu, G.; et al. Transcriptome Analysis and Gene Identification in the Pulmonary Artery of Broilers with Ascites Syndrome. PLoS ONE 2016, 11, e0156045. [Google Scholar] [CrossRef]
- Li, B.; Song, K.; Meng, J.; Li, L.; Zhang, G. Integrated application of transcriptomics and metabolomics provides insights into glycogen content regulation in the Pacific oyster Crassostrea gigas. BMC Genom. 2017, 18, 713. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatmary, A.C.; Nossal, R.; Parent, C.A.; Majumdar, R. Modeling neutrophil migration in dynamic chemoattractant gradients: Assessing the role of exosomes during signal relay. Mol. Biol. Cell 2017, 28, 3457–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. Pillars article: The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Fritz, J.H.; Kufer, T.A. Editorial: NLR-Protein Functions in Immunity. Front. Immunol. 2015, 6, 306. [Google Scholar] [CrossRef] [Green Version]
- Luangsay, S.; Ait-Goughoulte, M.; Michelet, M.; Floriot, O.; Bonnin, M.; Gruffaz, M.; Rivoire, M.; Fletcher, S.; Javanbakht, H.; Lucifora, J.; et al. Expression and functionality of toll-and RIG-like receptors in HepaRG cells. J. Hepatol. 2015, 63, 1077–1085. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Cheng, A.; Wang, M. Innate sensing of viruses by pattern recognition receptors in birds. Vet. Res. 2013, 44, 82. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wohlford-Lenane, C.; Zhao, J.; Fleming, E.; Lane, T.E.; McCray, P.B., Jr.; Perlman, S. Intranasal treatment with poly(I*C) protects aged mice from lethal respiratory virus infections. J. Virol. 2012, 86, 11416–11424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Bryan, R.; Scott, P.; Sydlaske, A.; Rose, D.M.; Terkeltaub, R. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2005, 52, 2936–2946. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Zamorano Cuervo, N.; Osseman, Q.; Grandvaux, N. Virus Infection Triggers MAVS Polymers of Distinct Molecular Weight. Viruses 2018, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Zalinger, Z.B.; Elliott, R.; Rose, K.M.; Weiss, S.R. MDA5 Is Critical to Host Defense during Infection with Murine Coronavirus. J. Virol. 2015, 89, 12330–12340. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [Green Version]
- Di Cara, F.; Sheshachalam, A.; Braverman, N.E.; Rachubinski, R.A.; Simmonds, A.J. Peroxisome-Mediated Metabolism Is Required for Immune Response to Microbial Infection. Immunity 2017, 47, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Limon-Pacheco, J.; Gonsebatt, M.E. The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutat. Res. 2009, 674, 137–147. [Google Scholar] [CrossRef]
- Smith, J.J.; Aitchison, J.D. Peroxisomes take shape. Nat. Rev. Mol. Cell Biol. 2013, 14, 803–817. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sahoo, M.; Lantier, L.; Warawa, J.; Cordero, H.; Deobald, K.; Re, F. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018, 14, e1007105. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Maesaka, J.K.; Fishbane, S. Regulation of renal urate excretion: A critical review. Am. J. Kidney Dis. 1998, 32, 917–933. [Google Scholar] [CrossRef]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1. [Google Scholar] [CrossRef]
- Taniguchi, K.; Tamura, Y.; Kumagai, T.; Shibata, S.; Uchida, S. Stimulation of V1a receptor increases renal uric acid clearance via urate transporters: Insight into pathogenesis of hypouricemia in SIADH. Clin. Exp. Nephrol. 2016, 20, 845–852. [Google Scholar] [CrossRef]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Lin, S.H.; Wang, Y.; Chin, Y.E.; Kang, L.; Mi, J. Glutamic Pyruvate Transaminase GPT2 Promotes Tumorigenesis of Breast Cancer Cells by Activating Sonic Hedgehog Signaling. Theranostics 2017, 7, 3021–3033. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Pang, S. Proline Catabolism Modulates Innate Immunity in Caenorhabditis elegans. Cell Rep. 2016, 17, 2837–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Yin, Y.L.; Li, D.; Kim, S.W.; Wu, G. Amino acids and immune function. Br. J. Nutr. 2007, 98, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Gu, C.; Ren, M.; Chen, D.; Yu, B.; He, J.; Yu, J.; Zheng, P.; Luo, J.; Luo, Y.; et al. l-Isoleucine Administration Alleviates Rotavirus Infection and Immune Response in the Weaned Piglet Model. Front. Immunol. 2018, 9, 1654. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Santiago, C.E.; Rivas-Santiago, B.; Leon, D.A.; Castaneda-Delgado, J.; Hernandez Pando, R. Induction of beta-defensins by l-isoleucine as novel immunotherapy in experimental murine tuberculosis. Clin. Exp. Immunol. 2011, 164, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Kakazu, E.; Kanno, N.; Ueno, Y.; Shimosegawa, T. Extracellular Branched-Chain Amino Acids, Especially Valine, Regulate Maturation and Function of Monocyte-Derived Dendritic Cells. J. Immunol. 2007, 179, 7137–7146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, D.; Demangel, C.; Munier-Jolain, K.; Miossec, S.; Geahel, I. Factors controlling cell proliferation and antibody production in mouse hybridoma cells: I. Influence of the amino acid supply. Biotechnol. Bioeng. 1991, 38, 561–570. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. MicrobiologyOpen 2018. [Google Scholar] [CrossRef]
- Lange, A.; Beier, S.; Steimle, A.; Autenrieth, I.B.; Huson, D.H.; Frick, J.-S. Extensive Mobilome-Driven Genome Diversification in Mouse Gut-AssociatedBacteroides vulgatusmpk. Genome Biol. Evol. 2016, 8, 1197–1207. [Google Scholar] [CrossRef] [Green Version]
- Maerz, J.K.; Steimle, A.; Lange, A.; Bender, A.; Fehrenbacher, B.; Frick, J.-S. Outer membrane vesicles blebbing contributes to B. vulgatus mpk-mediated immune response silencing. Gut Microbes 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, N.; Iwamoto, C.; Kano, H.; Yamaoka, N.; Fukuuchi, T.; Kaneko, K.; Asami, Y. Evaluation of purine utilization by Lactobacillus gasseri strains with potential to decrease the absorption of food-derived purines in the human intestine. Nucleosides Nucleotides Nucleic Acids 2016, 35, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Liu, P.; Zhou, C.; Shi, Y.; Wu, Q.; Yang, Y.; Li, G.; Hu, G.; Guo, X. A Multi-Omics Study of Chicken Infected by Nephropathogenic Infectious Bronchitis Virus. Viruses 2019, 11, 1070. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111070
Xu P, Liu P, Zhou C, Shi Y, Wu Q, Yang Y, Li G, Hu G, Guo X. A Multi-Omics Study of Chicken Infected by Nephropathogenic Infectious Bronchitis Virus. Viruses. 2019; 11(11):1070. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111070
Chicago/Turabian StyleXu, Puzhi, Ping Liu, Changming Zhou, Yan Shi, Qingpeng Wu, Yitian Yang, Guyue Li, Guoliang Hu, and Xiaoquan Guo. 2019. "A Multi-Omics Study of Chicken Infected by Nephropathogenic Infectious Bronchitis Virus" Viruses 11, no. 11: 1070. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111070