New Genus Fibralongavirus in Siphoviridae Phages of Staphylococcus pseudintermedius

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Bacteriophages
2.2. Growth Properties
2.3. Phage Propagation and Purification
2.4. Electron and Cryo-Electron Microscopy
2.5. DNA Extraction from the Phage Particles
2.6. Partial rpoB Gene Sequencing and Phylogenetic Analysis
2.7. Genome Sequencing and Bioinformatic Analyses
2.8. Pulsed-Field Gel Electrophoresis.
2.9. SDS-PAGE of Structural Proteins
2.10. Filter-Aided Sample Preparation for Mass Spectrometry and LC-MS/MS Analysis
2.11. Nucleotide Sequence Accession Numbers
3. Results
3.1. Growth Characteristics of Phage QT1
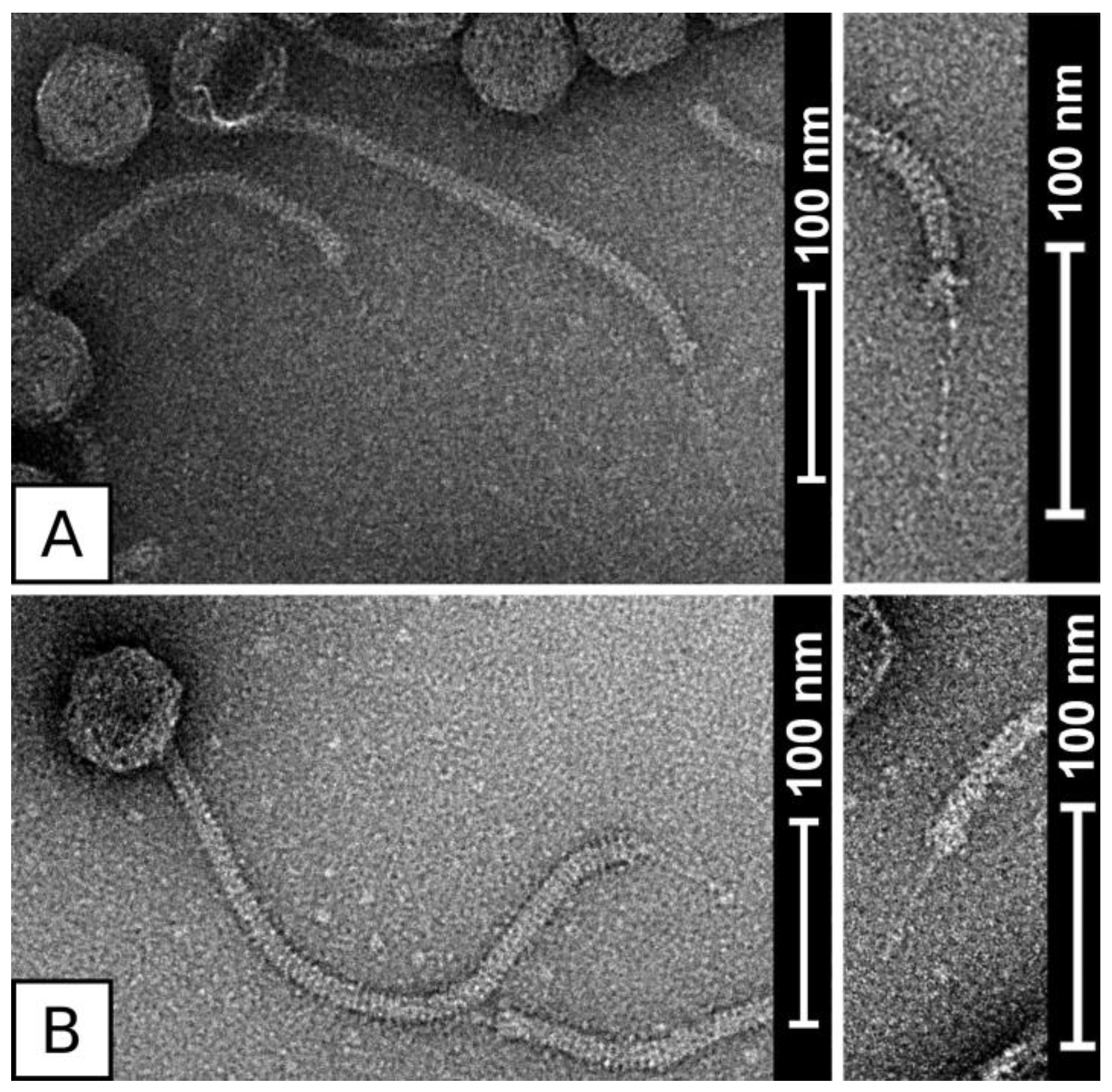
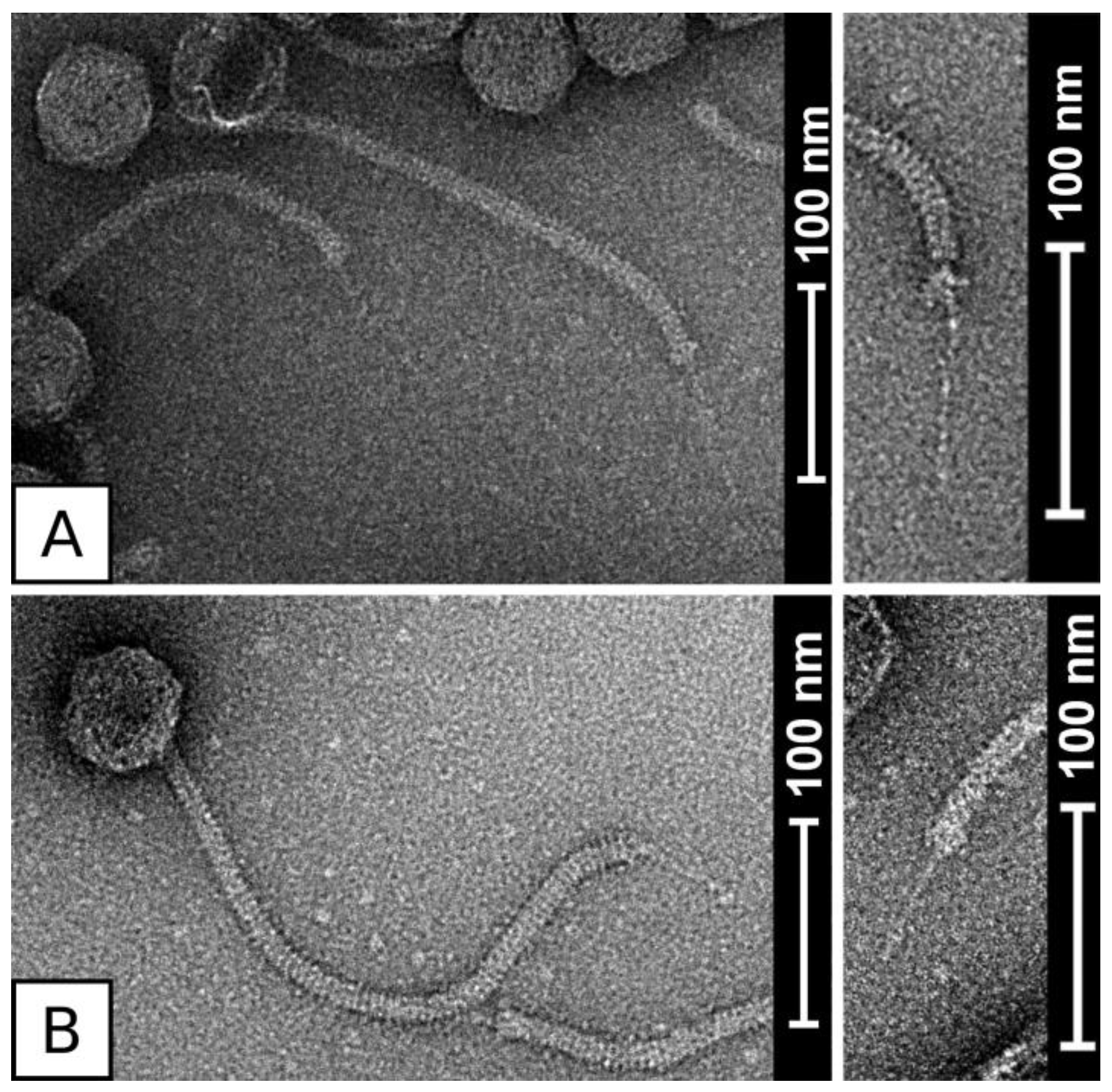
3.2. Virion Morphology of Phages QT1 and 2638A
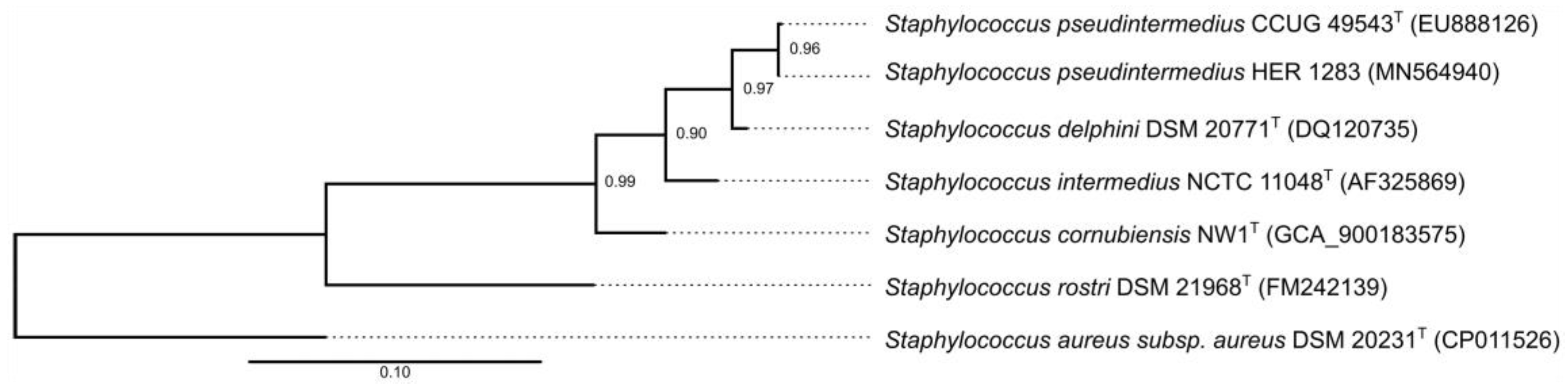
3.3. Reclassification of the Propagating Strain Staphylococcus aureus 2854 for Phage 2638A as Staphylococcus pseudintermedius and Phage Host Range
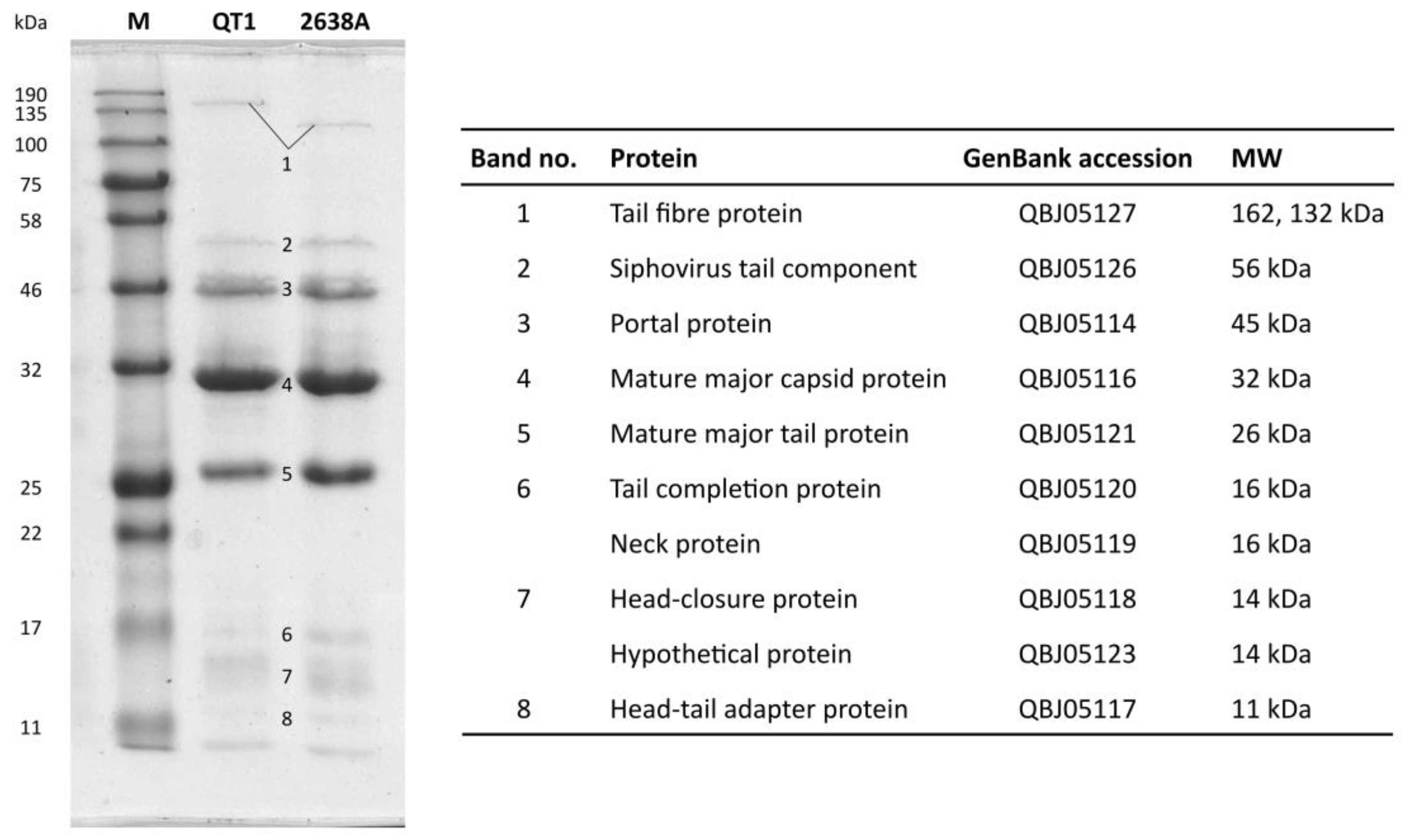
3.4. Phage Structural Proteins
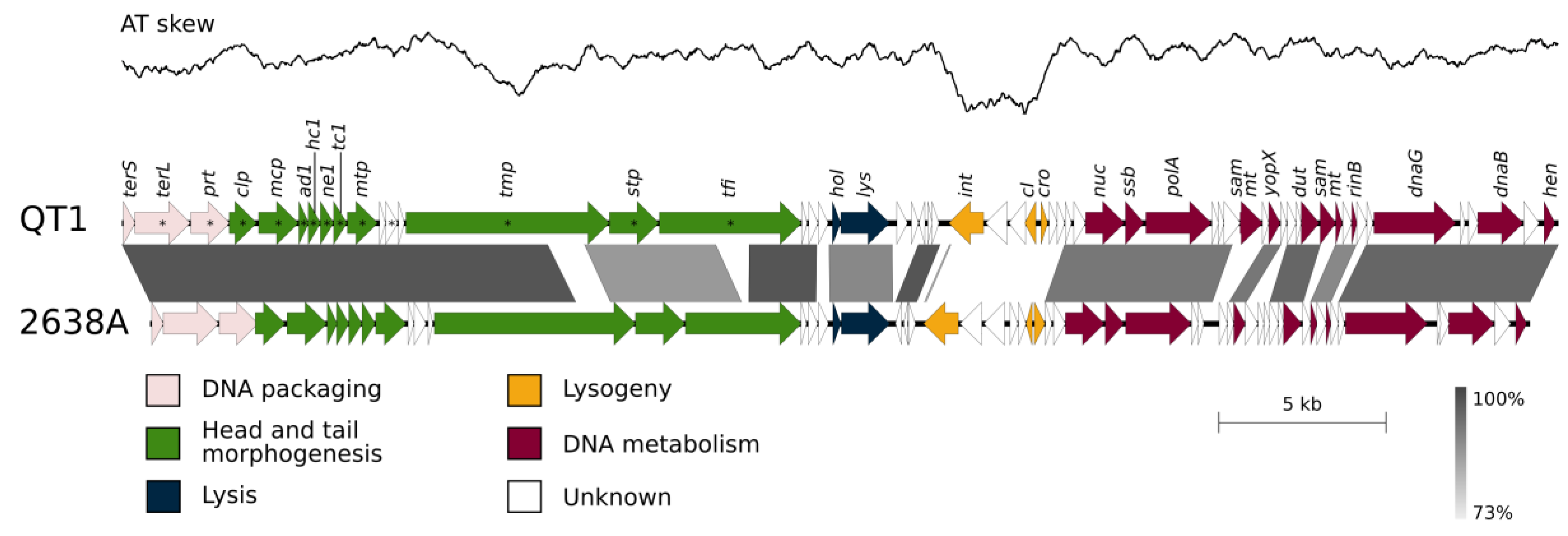
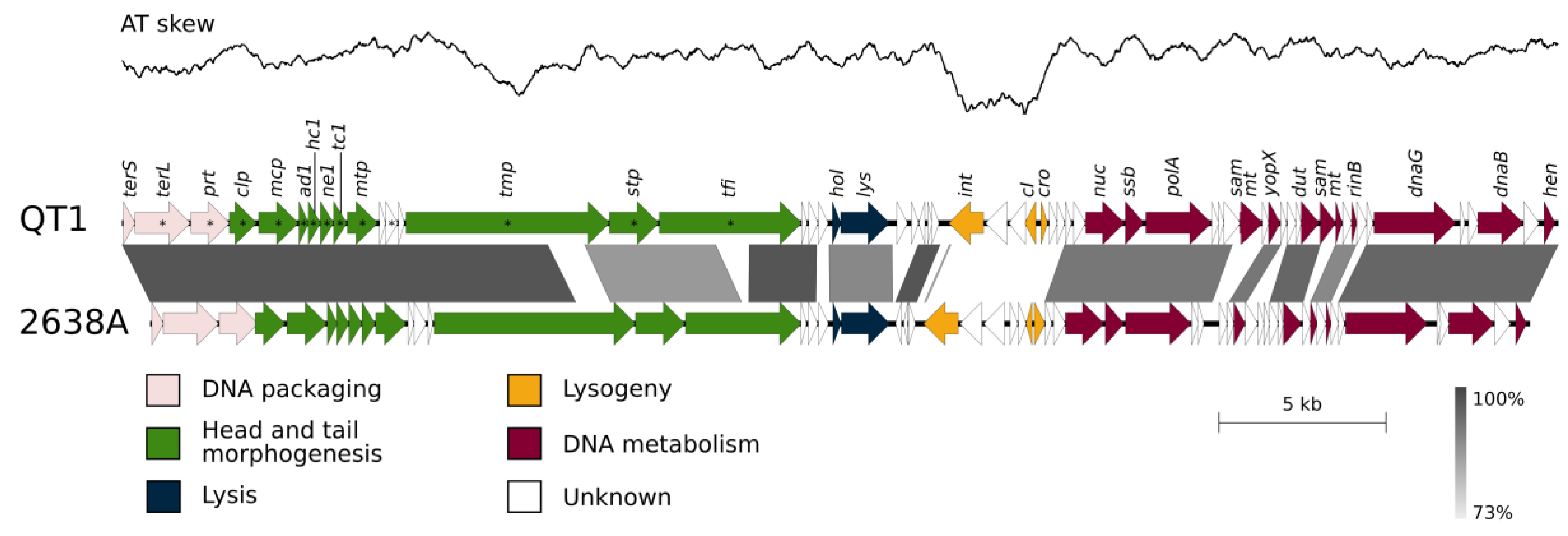
3.5. QT1 Genome Description
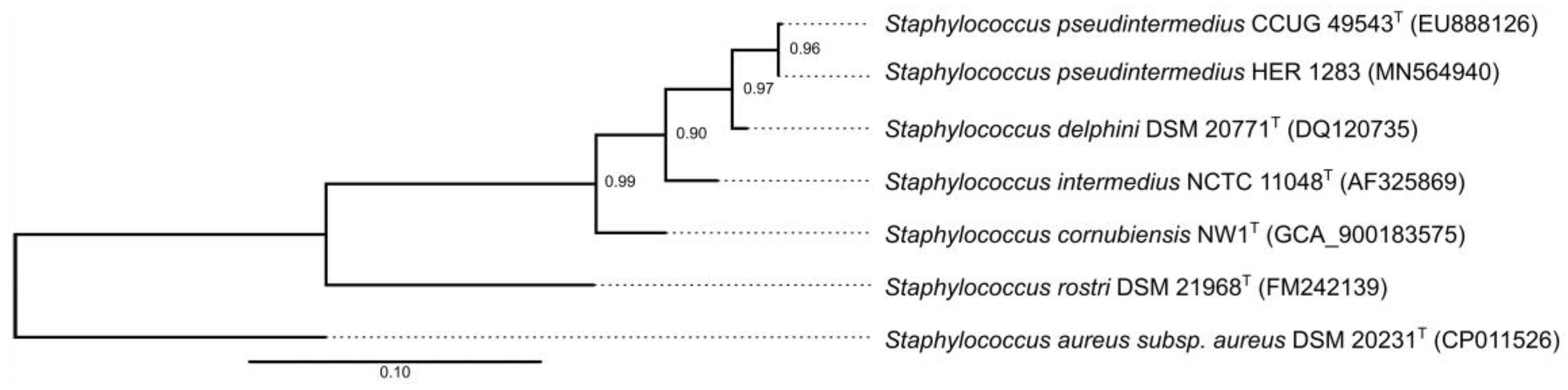
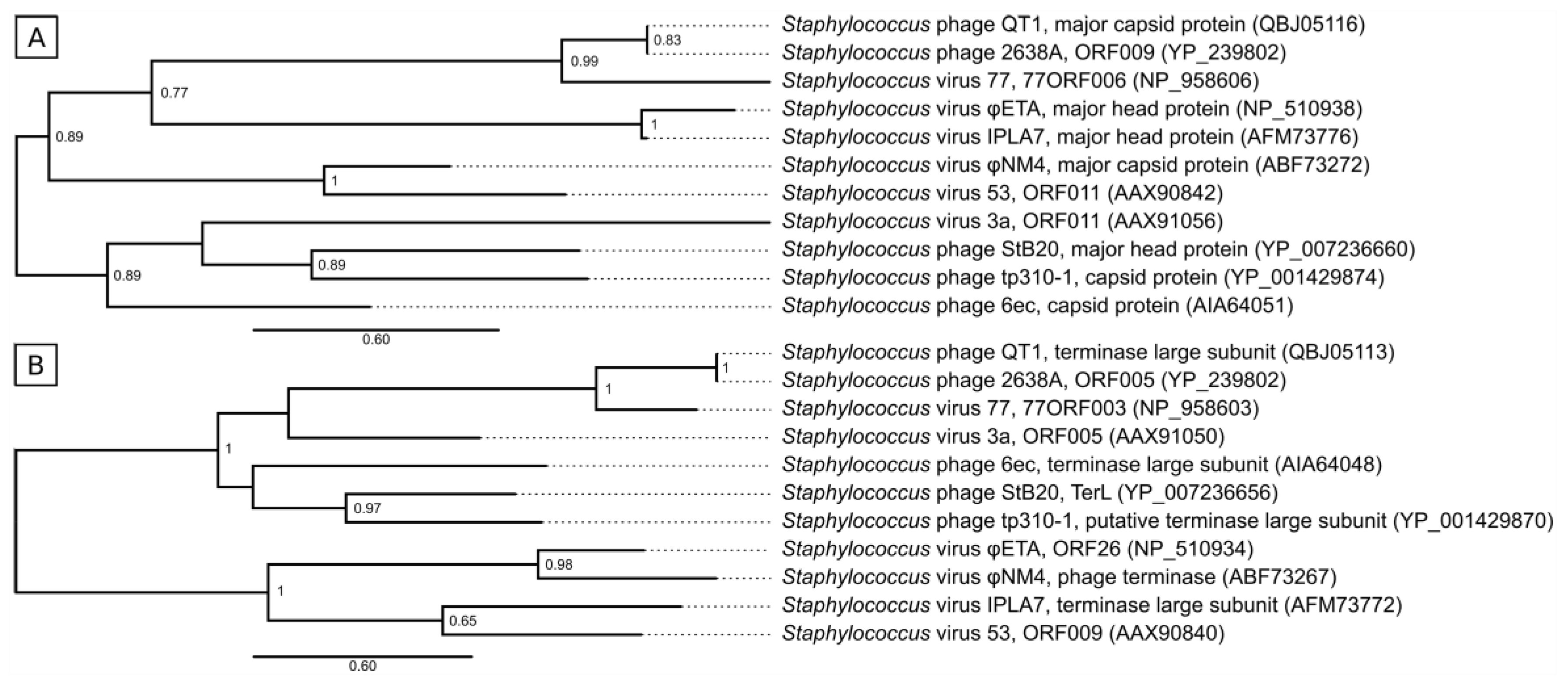
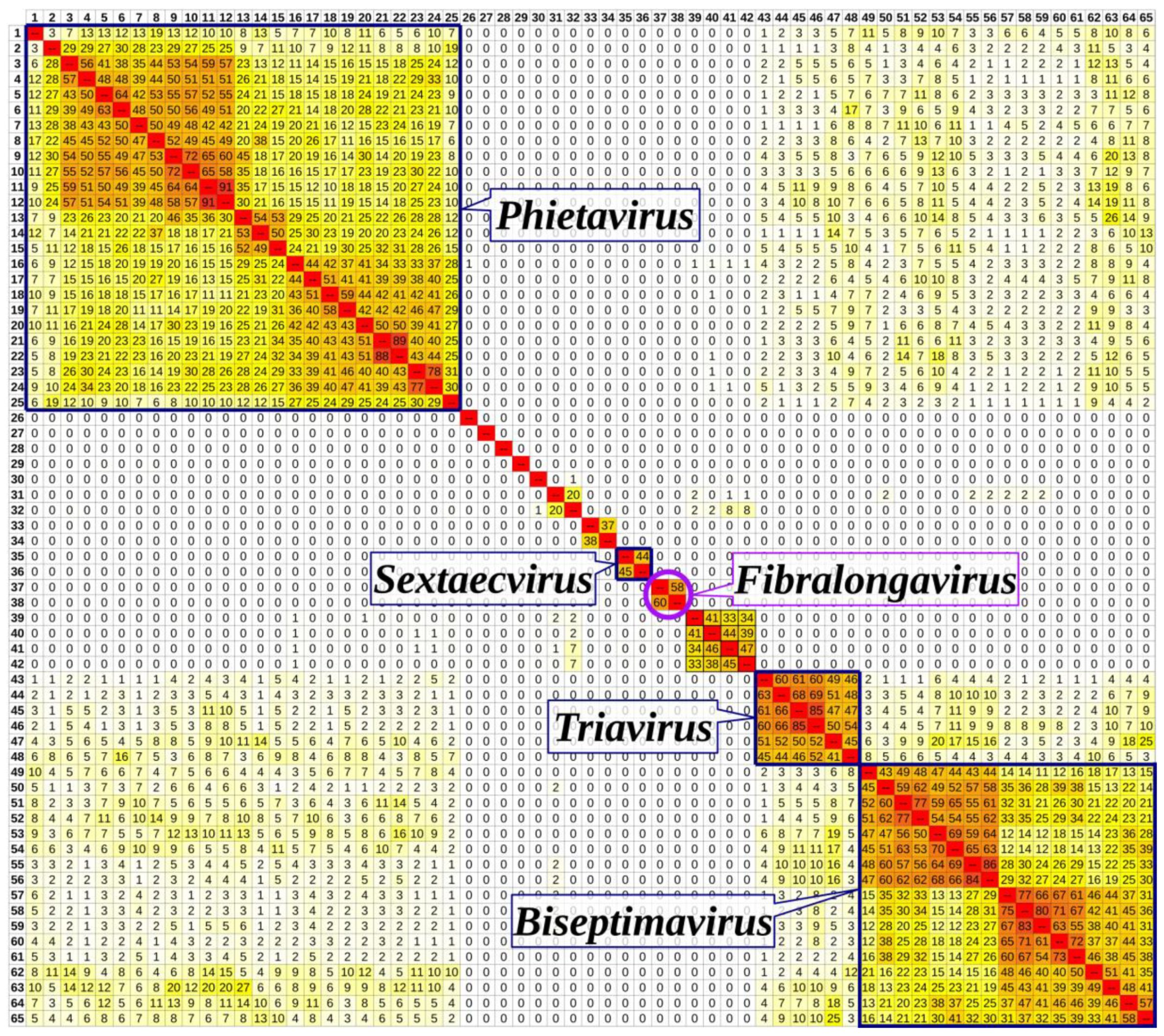
3.6. Taxonomical Classification and Proposal of New Genus Fibralongavirus
3.7. Genome Description of Propagating Strain for Phage QT1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sasaki, T.; Kikuchi, K.; Tanaka, Y.; Takahashi, N.; Kamata, S.; Hiramatsu, K. Reclassification of phenotypically identified Staphylococcus intermedius strains. J. Clin. Microbiol. 2007, 45, 2770–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannoehr, J.; Franco, A.; Iurescia, M.; Battisti, A.; Fitzgerald, J.R. Molecular diagnostic identification of Staphylococcus pseudintermedius. J. Clin. Microbiol. 2009, 47, 469–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Švec, P.; Pantůček, R.; Petráš, P.; Sedláček, I.; Nováková, D. Identification of Staphylococcus spp. using (GTG)5-PCR fingerprinting. Syst. Appl. Microbiol. 2010, 33, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Bannoehr, J.; Ben Zakour, N.L.; Waller, A.S.; Guardabassi, L.; Thoday, K.L.; van den Broek, A.H.; Fitzgerald, J.R. Population genetic structure of the Staphylococcus intermedius group: Insights into agr diversification and the emergence of methicillin-resistant strains. J. Bacteriol. 2007, 189, 8685–8692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannoehr, J.; Guardabassi, L. Staphylococcus pseudintermedius in the dog: Taxonomy, diagnostics, ecology, epidemiology and pathogenicity. Vet. Dermatol. 2012, 23, 253-e52. [Google Scholar] [CrossRef] [PubMed]
- Börjesson, S.; Gómez-Sanz, E.; Ekström, K.; Torres, C.; Grönlund, U. Staphylococcus pseudintermedius can be misdiagnosed as Staphylococcus aureus in humans with dog bite wounds. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 34, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Riegel, P.; Jesel-Morel, L.; Laventie, B.; Boisset, S.; Vandenesch, F.; Prévost, G. Coagulase-positive Staphylococcus pseudintermedius from animals causing human endocarditis. Int. J. Med. Microbiol. 2011, 301, 237–239. [Google Scholar] [CrossRef]
- Van Duijkeren, E.; Catry, B.; Greko, C.; Moreno, M.A.; Pomba, M.C.; Pyorala, S.; Ruzauskas, M.; Sanders, P.; Threlfall, E.J.; Torren-Edo, J.; et al. Review on methicillin-resistant Staphylococcus pseudintermedius. J. Antimicrob. Chemother. 2011, 66, 2705–2714. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency (EMA). Reflection Paper on Meticillin-Resistant Staphylococcus pseudintermedius EMEA/CVMP/SAGAM/736964/2009. Available online: https://www.ema.europa.eu/en/meticillin-resistant-staphylococcus-pseudintermedius (accessed on 13 November 2019).
- Kawano, J.; Shimizu, A.; Kimura, S.; Blouse, L. Experimental bacteriophage set for typing Staphylococcus intermedius. Zentralbl. Bakteriol. Mikrobiol. Hyg. A 1982, 253, 321–330. [Google Scholar] [CrossRef]
- Overturf, G.D.; Talan, D.A.; Singer, K.; Anderson, N.; Miller, J.I.; Greene, R.T.; Froman, S. Phage typing of Staphylococcus intermedius. J. Clin. Microbiol. 1991, 29, 373–375. [Google Scholar]
- Wakita, Y.; Shimizu, A.; Hájek, V.; Kawano, J.; Yamashita, K. Characterization of Staphylococcus intermedius from pigeons, dogs, foxes, mink, and horses by pulsed-field gel electrophoresis. J. Vet. Med. Sci. 2002, 64, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of vB_SauM-fRuSau02, a Twort-like bacteriophage isolated from a therapeutic phage cocktail. Viruses 2017, 9, 258. [Google Scholar] [CrossRef] [PubMed]
- Moodley, A.; Kot, W.; Nälgård, S.; Jakociune, D.; Neve, H.; Hansen, L.H.; Guardabassi, L.; Vogensen, F.K. Isolation and characterization of bacteriophages active against methicillin-resistant Staphylococcus pseudintermedius. Res. Vet. Sci. 2019, 122, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Melter, O.; Švec, P.; Tkadlec, J.; Doškař, J.; Kinská, H.; Pantůček, R. Characterisation of methicillin-susceptible Staphylococcus pseudintermedius isolates from canine infections and determination of virulence factors using multiplex PCR. Vet. Med. 2017, 62, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Mališová, L.; Šafránková, R.; Kekláková, J.; Petráš, P.; Žemličková, H.; Jakubů, V. Correct species identification (reclassification in CNCTC) of strains of Staphylococcus intermedius-group can improve an insight into their evolutionary history. Folia Microbiol. 2018, 64, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Practical Methods for Determining Phage Growth Parameters. In Bacteriophages. Methods in Molecular Biology; Clokie, M.R., Kropinski, A.M., Eds.; Humana Press: New York, NY, USA, 2009; Volume 501, pp. 175–202. [Google Scholar] [CrossRef]
- Mašlaňová, I.; Stříbná, S.; Doškař, J.; Pantůček, R. Efficient plasmid transduction to Staphylococcus aureus strains insensitive to the lytic action of transducing phage. FEMS Microbiol. Lett. 2016, 363, fnw211. [Google Scholar] [CrossRef] [PubMed]
- Subcommittee on Taxonomy of Staphylococci and Micrococci. Recommendations. Int. Bull. Bacteriol. Nomencl. Taxon. 1965, 15, 109–110. [Google Scholar] [CrossRef]
- Cadness-Graves, B.; Williams, R.; Harper, G.J.; Miles, A.A. Slide-test for coagulase-positive staphylococci. Lancet 1943, 241, 736–738. [Google Scholar] [CrossRef]
- Mašlaňová, I.; Doškař, J.; Varga, M.; Kuntová, L.; Mužík, J.; Malúšková, D.; Růžičková, V.; Pantůček, R. Bacteriophages of Staphylococcus aureus efficiently package various bacterial genes and mobile genetic elements including SCCmec with different frequencies. Environ. Microbiol. Rep. 2013, 5, 66–73. [Google Scholar] [CrossRef]
- Mellmann, A.; Becker, K.; von Eiff, C.; Keckevoet, U.; Schumann, P.; Harmsen, D. Sequencing and staphylococci identification. Emerging Infect. Dis. 2006, 12, 333–336. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics, Babraham Institute, Cambridge, UK. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 June 2018).
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling Genomes and Mini-Metagenomes from Highly Chimeric Reads. In Research in Computational Molecular Biology; Minghua, D., Rui, J., Fengzhu, S., Xuegong, Z., Eds.; Springer: Berlin, Germany, 2013; Volume 7821, pp. 158–170. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, E.W.; Miller, W. Optimal alignments in linear space. Comput. Appl. Biosci. 1988, 4, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999, 27, 3911–3920. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Abricate: Mass Screening of Contigs for Antimicrobial and Virulence Genes. Department of Microbiology and Immunology, The University of Melbourne, Melbourne, Australia. Available online: https://github.com/tseemann/abricate (accessed on 28 February 2019).
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.-H.; McDermott, P.F.; et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific a-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A. FigTree: Produce High-Quality Figures of Phylogenetic Trees. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, UK. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 9 August 2019).
- Ahmed, N.; Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef]
- Candiano, G.; Bruschi, M.; Musante, L.; Santucci, L.; Ghiggeri, G.M.; Carnemolla, B.; Orecchia, P.; Zardi, L.; Righetti, P.G. Blue silver: A very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 2004, 25, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Ostasiewicz, P.; Mann, M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 2011, 10, 3040–3049. [Google Scholar] [CrossRef]
- Stejskal, K.; Potěšil, D.; Zdráhal, Z. Suppression of peptide sample losses in autosampler vials. J. Proteome Res. 2013, 12, 3057–3062. [Google Scholar] [CrossRef]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [Green Version]
- Slopek, S.; Krzywy, T. Morphology and ultrastructure of bacteriophages. An electron microscopic study. Arch. Immunol. Ther. Exp. 1985, 33, 12–17. [Google Scholar]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.C.; Dickmanns, A.; Urlaub, H.; Schmitt, A.; Mühlenhoff, M.; Stummeyer, K.; Schwarzer, D.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of an intramolecular chaperone mediating triple-β-helix folding. Nat. Struct. Mol. Biol. 2010, 17, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Wipf, J.R.K.; Deutsch, D.R.; Westblade, L.F.; Fischetti, V.A.; Putonti, C. Genome sequences of six prophages isolated from Staphylococcus pseudintermedius strains recovered from human and animal clinical specimens. Microbiol. Res. Announc. 2019, 8, e00387-19. [Google Scholar] [CrossRef] [Green Version]
- Nowakowski, M.; Jaremko, Ł.; Wladyka, B.; Dubin, G.; Ejchart, A.; Mak, P. Spatial attributes of the four-helix bundle group of bacteriocins – The high-resolution structure of BacSp222 in solution. Int. J. Biol. Macromol. 2018, 107, 2715–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.H.; Hwang, C.Y. First detection of multiresistance pRE25-like elements from Enterococcus spp. in Staphylococcus pseudintermedius isolated from canine pyoderma. J. Glob. Antimicrob. Resist. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Drancourt, M.; Raoult, D. rpoB gene sequence-based identification of Staphylococcus species. J. Clin. Microbiol. 2002, 40, 1333–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, A.H.; Kadoi, K.; Miyanaga, K.; Usui, M.; Tamura, Y.; Cui, L.; Tanji, Y. Analysis host-recognition mechanism of staphylococcal kayvirus φSA039 reveals a novel strategy that protects Staphylococcus aureus against infection by Staphylococcus pseudintermedius Siphoviridae phages. Appl. Microbiol. Biotechnol. 2019, 103, 6809–6823. [Google Scholar] [CrossRef] [PubMed]
- Ben Zakour, N.L.; Beatson, S.A.; van den Broek, A.H.M.; Thoday, K.L.; Fitzgerald, J.R. Comparative genomics of the Staphylococcus intermedius group of animal pathogens. Front. Cell. Infect. Microbiol. 2012, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T. Lysis from without. Bacteriophage 2014, 1, 46–49. [Google Scholar] [CrossRef]
- Ingmer, H.; Gerlach, D.; Wolz, C. Temperate phages of Staphylococcus aureus. Microbiol. Spectrum 2019, 7. [Google Scholar] [CrossRef]
- Brüssow, H.; Desiere, F. Comparative phage genomics and the evolution of Siphoviridae: Insights from dairy phages. Mol. Microbiol. 2001, 39, 213–223. [Google Scholar] [CrossRef]
- Quiles-Puchalt, N.; Carpena, N.; Alonso, J.C.; Novick, R.P.; Marina, A.; Penadés, J.R. Staphylococcal pathogenicity island DNA packaging system involving cos-site packaging and phage-encoded HNH endonucleases. Proc. Natl Acad. Sci. USA 2014, 111, 6016–6021. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.; Verstappen, K.M.; Huijbregts, L.; Spaninks, M.; Wagenaar, J.A.; Fluit, A.C.; Duim, B. Development of a real-time PCR for detection of Staphylococcus pseudintermedius using a novel automated comparison of whole-genome sequences. PLoS ONE 2017, 12, e0183925. [Google Scholar] [CrossRef] [Green Version]
- Cernooka, E.; Rumnieks, J.; Tars, K.; Kazaks, A. Structural basis for DNA recognition of a single-stranded DNA-binding protein from Enterobacter phage Enc34. Sci. Rep. 2017, 7, 15529. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, R.M. Sau42I, a II-like restriction-modification system encoded by the Staphylococcus aureus quadruple-converting phage φ42. Microbiology 2005, 151, 1301–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, G.E.; Matthews, A.M.; King, D.G.; Lane, K.D.; Olivarez, N.P.; Tallent, S.M.; Gill, S.R.; Novick, R.P. The complete genomes of Staphylococcus aureus bacteriophages 80 and 80α—Implications for the specificity of SaPI mobilization. Virology 2010, 407, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossykh, V.G.; Schlagman, S.L.; Hattman, S. Phage T4 DNA [N]-adenine6 methyltransferase. Overexpression, purification, and characterization. J. Biol. Chem. 1995, 270, 14389–14393. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Chaperone–protein interactions that mediate assembly of the bacteriophage λ tail to the correct length. J. Mol. Biol. 2014, 426, 1004–1018. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rubio, L.; Gutiérrez, D.; Martínez, B.; Rodríguez, A.; Götz, F.; García, P. The tape measure protein of the Staphylococcus aureus bacteriophage vB_SauS-φIPLA35 has an active muramidase domain. Appl. Environ. Microbiol. 2012, 78, 6369–6371. [Google Scholar] [CrossRef] [Green Version]
- North, O.I.; Sakai, K.; Yamashita, E.; Nakagawa, A.; Iwazaki, T.; Buttner, C.R.; Takeda, S.; Davidson, A.R. Phage tail fibre assembly proteins employ a modular structure to drive the correct folding of diverse fibres. Nat. Microbiol. 2019, 4, 1645–1653. [Google Scholar] [CrossRef]
- Neethirajan, S.; DiCicco, M. Atomic force microscopy study of the antibacterial effect of fosfomycin on methicillin-resistant Staphylococcus pseudintermedius. Appl. Nanosci. 2013, 4, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Schuch, R.; Fischetti, V.A. Detailed genomic analysis of the Wβ and γ phages infecting Bacillus anthracis: Implications for evolution of environmental fitness and antibiotic resistance. J. Bacteriol. 2006, 188, 3037–3051. [Google Scholar] [CrossRef] [Green Version]
- Schade, S.Z.; Adler, J.; Ris, H. How bacteriophage χ attacks motile bacteria. J. Virol. 1967, 1, 599–609. [Google Scholar]
- Abaev, I.; Foster-Frey, J.; Korobova, O.; Shishkova, N.; Kiseleva, N.; Kopylov, P.; Pryamchuk, S.; Schmelcher, M.; Becker, S.C.; Donovan, D.M. Staphylococcal phage 2638A endolysin is lytic for Staphylococcus aureus and harbors an inter-lytic-domain secondary translational start site. Appl. Microbiol. Biotechnol. 2012, 97, 3449–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, S.C.; Foster-Frey, J.; Stodola, A.J.; Anacker, D.; Donovan, D.M. Differentially conserved staphylococcal SH3b_5 cell wall binding domains confer increased staphylolytic and streptolytic activity to a streptococcal prophage endolysin domain. Gene 2009, 443, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Shen, Y.; Nelson, D.C.; Eugster, M.R.; Eichenseher, F.; Hanke, D.C.; Loessner, M.J.; Dong, S.; Pritchard, D.G.; Lee, J.C.; et al. Evolutionarily distinct bacteriophage endolysins featuring conserved peptidoglycan cleavage sites protect mice from MRSA infection. J. Antimicrob. Chemother. 2015, 70, 1453–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalskaya, N.Y.; Herndon, E.E.; Foster-Frey, J.A.; Donovan, D.M.; Hammond, R.W. Antimicrobial activity of bacteriophage derived triple fusion protein against Staphylococcus aureus. AIMS Microbiol. 2019, 5, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Daniel, A.; Bonnen, P.E.; Fischetti, V.A. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J. Bacteriol. 2006, 189, 2086–2100. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, D.; Martínez, B.; Rodríguez, A.; García, P. Genomic characterization of two Staphylococcus epidermidis bacteriophages with anti-biofilm potential. BMC Genom. 2012, 13, 228. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.; Gutiérrez, D.; García, P.; Rodríguez, A. The perfect bacteriophage for therapeutic applications—A quick guide. Antibiotics 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeman, M.; Bárdy, P.; Vrbovská, V.; Roudnický, P.; Zdráhal, Z.; Růžičková, V.; Doškař, J.; Pantůček, R. New Genus Fibralongavirus in Siphoviridae Phages of Staphylococcus pseudintermedius. Viruses 2019, 11, 1143. https://0-doi-org.brum.beds.ac.uk/10.3390/v11121143
Zeman M, Bárdy P, Vrbovská V, Roudnický P, Zdráhal Z, Růžičková V, Doškař J, Pantůček R. New Genus Fibralongavirus in Siphoviridae Phages of Staphylococcus pseudintermedius. Viruses. 2019; 11(12):1143. https://0-doi-org.brum.beds.ac.uk/10.3390/v11121143
Chicago/Turabian StyleZeman, Michal, Pavol Bárdy, Veronika Vrbovská, Pavel Roudnický, Zbyněk Zdráhal, Vladislava Růžičková, Jiří Doškař, and Roman Pantůček. 2019. "New Genus Fibralongavirus in Siphoviridae Phages of Staphylococcus pseudintermedius" Viruses 11, no. 12: 1143. https://0-doi-org.brum.beds.ac.uk/10.3390/v11121143