Prion Variants of Yeast are Numerous, Mutable, and Segregate on Growth, Affecting Prion Pathogenesis, Transmission Barriers, and Sensitivity to Anti-Prion Systems

Abstract
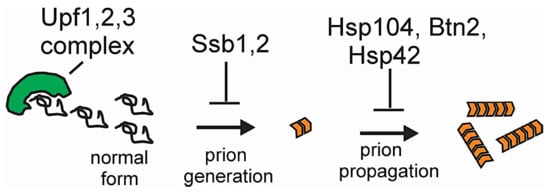
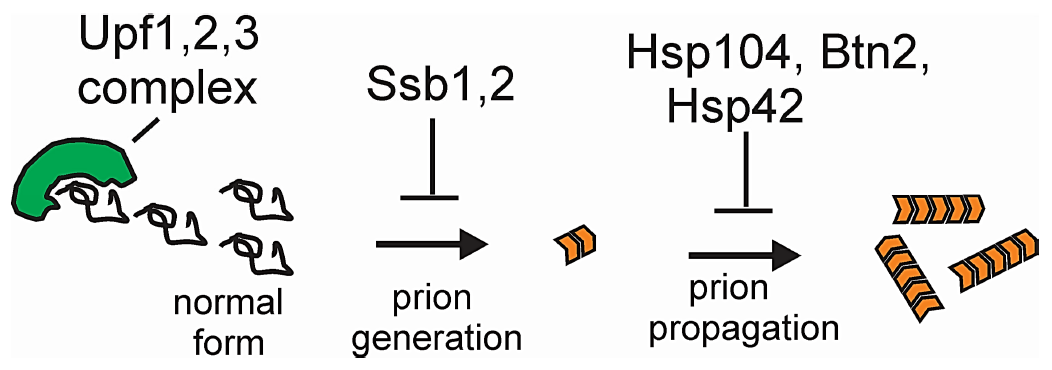
:
1. Introduction
2. Prion Variants/Strains
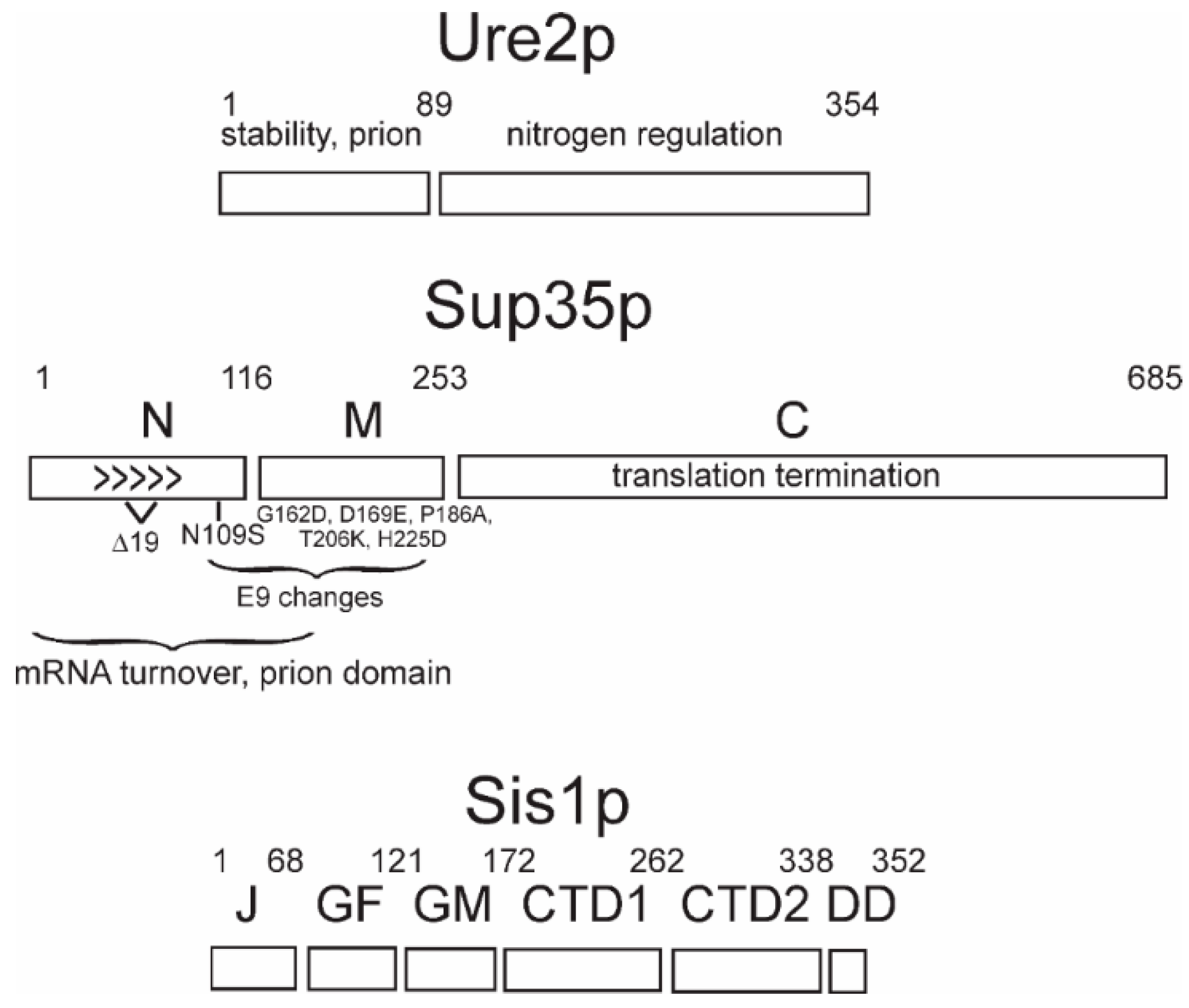
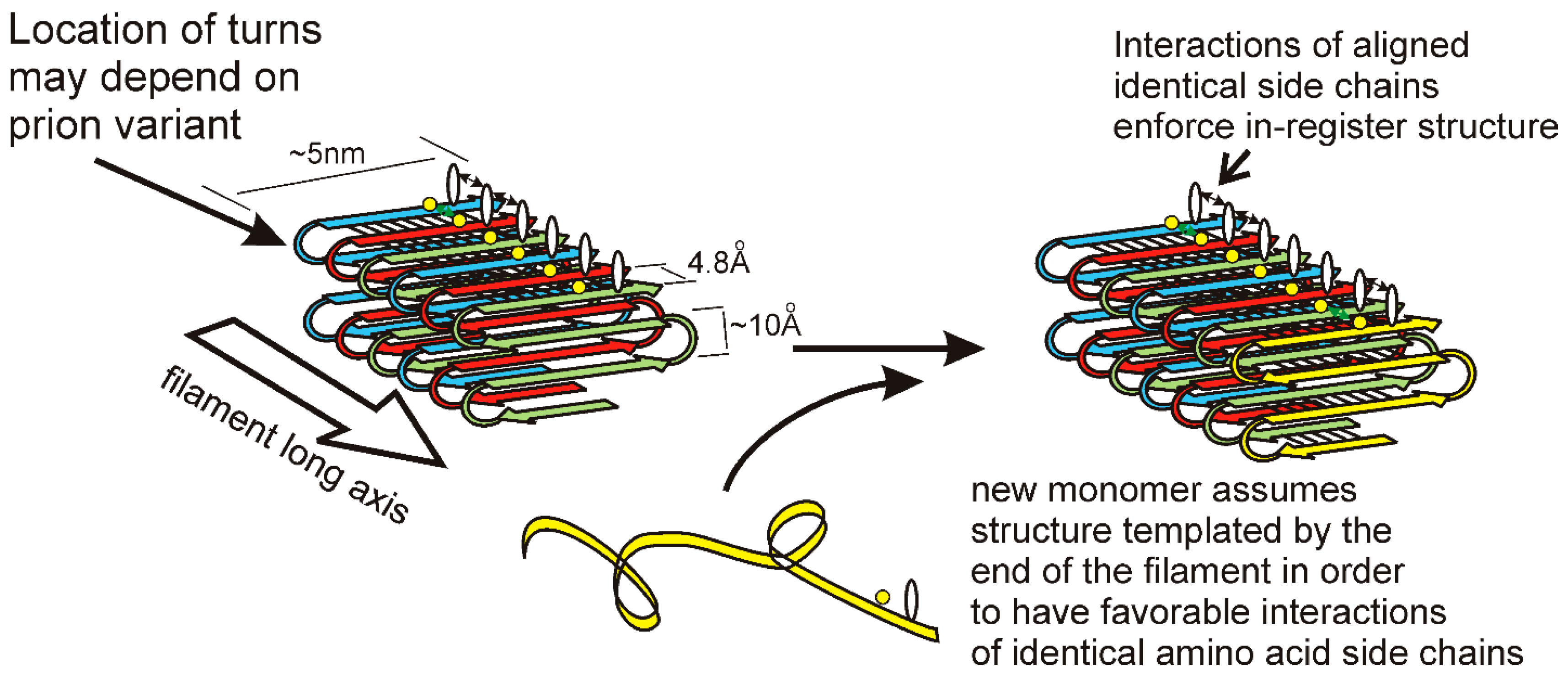
3. Prion Domains, Amyloid Architecture, and Propagation Mechanism
4. Detrimental Prions Have Variants, but Not the Beneficial [Het-s] Prion
5. Degree of Pathogenicity Varies with Prion Variant
6. Interspecies Transmission Barriers Vary with Prion Variant
7. Intraspecies Transmission Barriers and Prion Variants
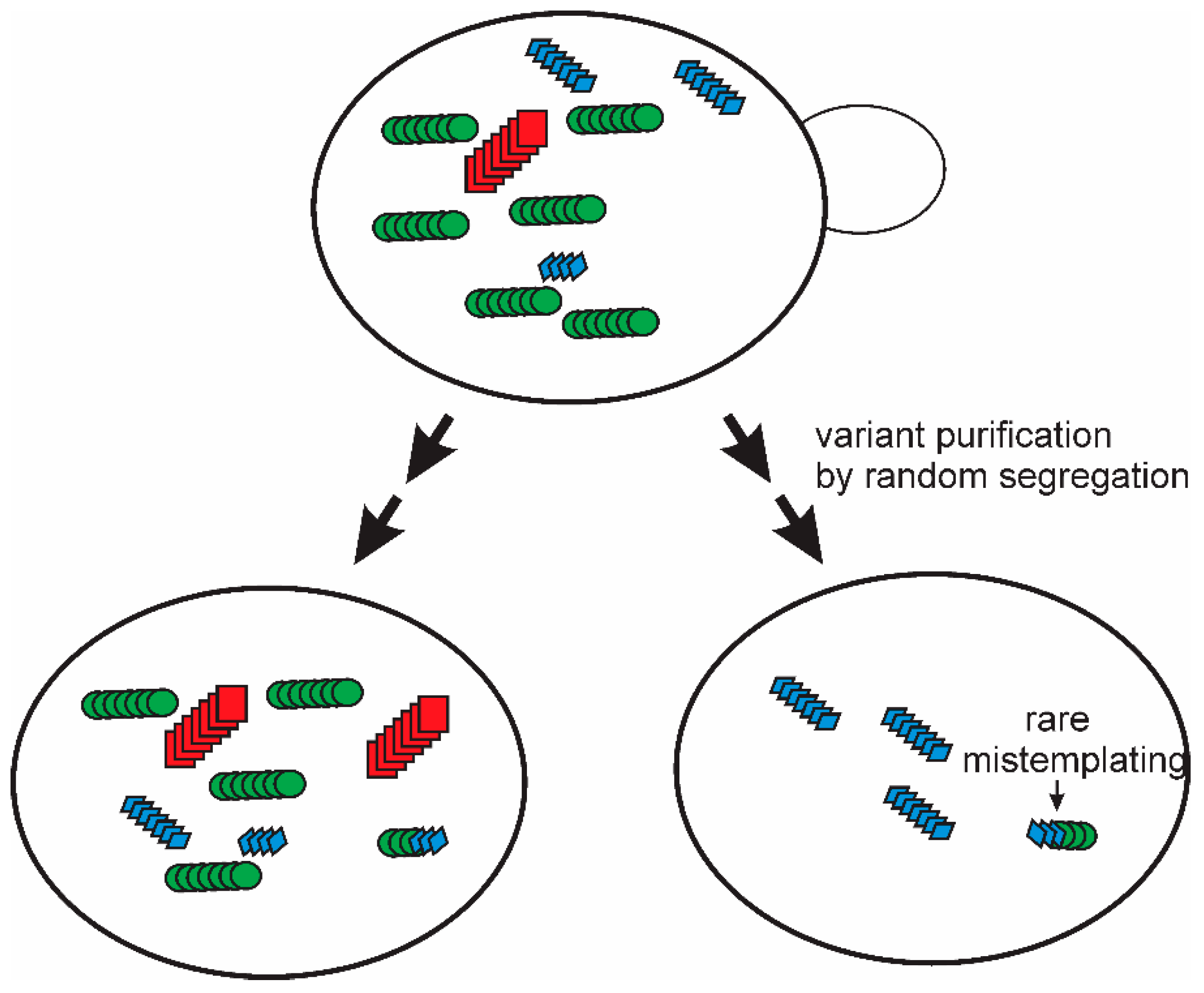
8. Prion Mutation and Segregation of Variants
9. Prion Variant Generation
10. Effects of Chaperones and Other Proteins on Prions
11. Anti-Prion Systems Normally Block Almost All Prion Variants from Appearing
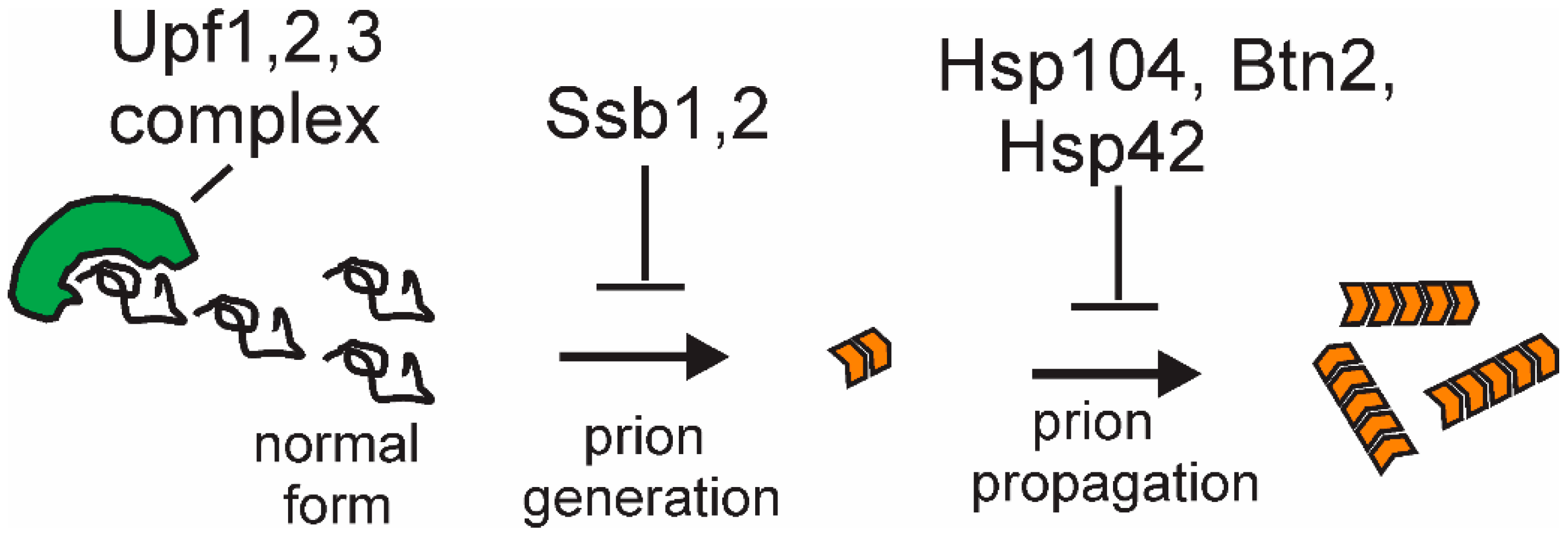
11.1. Ssb Ribosome-Associated Hsp70s Block Prion Formation
11.2. Normal Levels of Btn2p and Cur1p Cure the [URE3] Variants with Low Seed Number
11.3. Normal Levels of Hsp104 Cull Many [PSI+] Prion Variants
11.4. Normal Levels of Upf Proteins Cure Most Spontaneous Variants of [PSI+]
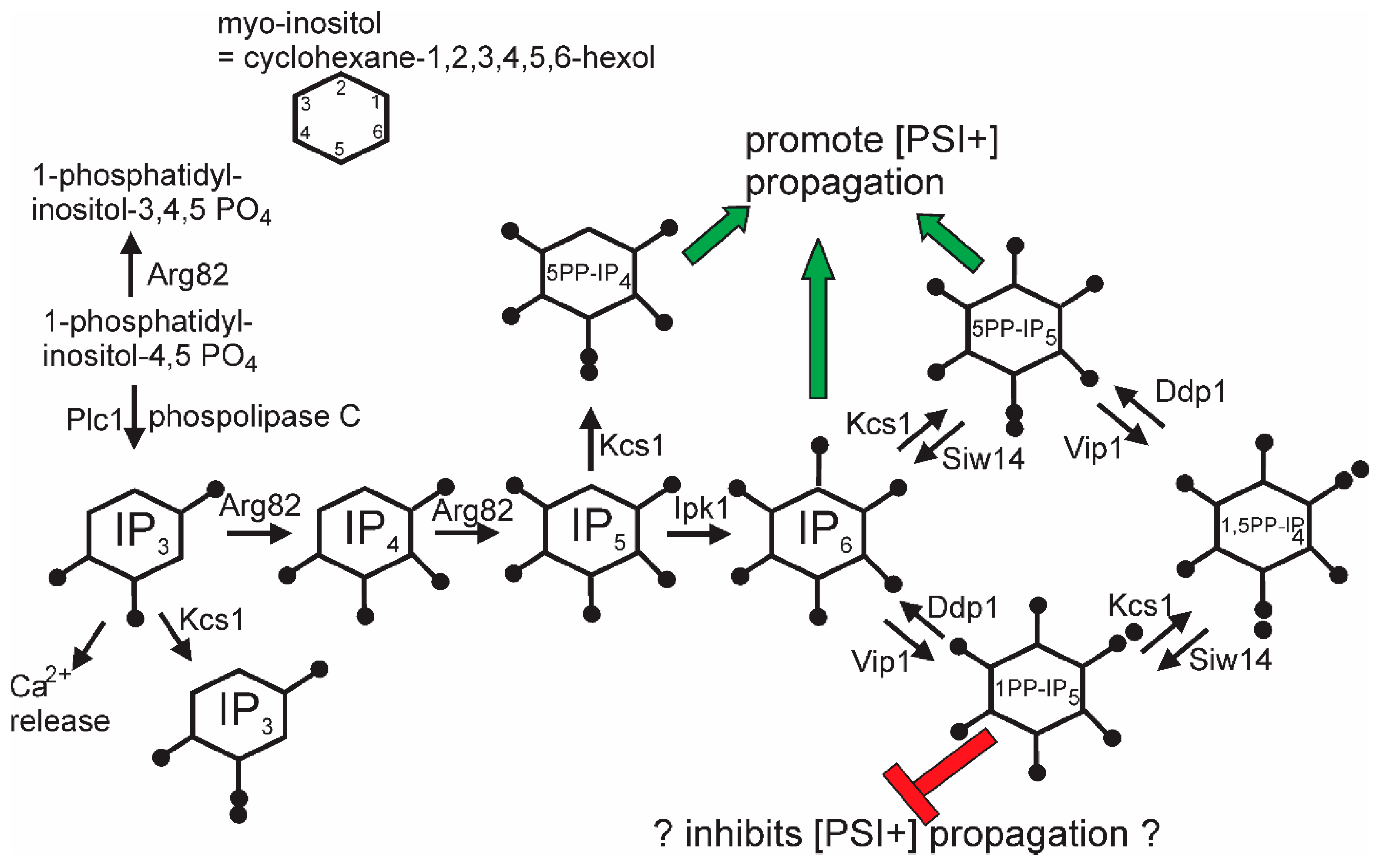
11.5. Inositol Poly/Pyro-Phosphates Involvement in [PSI+] Prion Propagation
12. Differential Effects of Chaperones on Prion Variants
13. The Chaperone Environment Selects Prion Variants
14. Prions Are More Abundant and Varied than Was Previously Thought
15. Implications for Human Disease
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffith, J.S. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in S. cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. (Ed.) Prion Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; p. 456. [Google Scholar]
- Roberts, B.T.; Wickner, R.B. A class of prions that propagate via covalent auto-activation. Genes Dev. 2003, 17, 2083–2087. [Google Scholar] [CrossRef] [PubMed]
- Lacroute, F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 1971, 106, 519–522. [Google Scholar] [PubMed]
- Masison, D.C.; Wickner, R.B. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995, 270, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Edskes, H.K.; Gray, V.T.; Wickner, R.B. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc. Natl. Acad. Sci. USA 1999, 96, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.L.; Cheng, N.; Williams, R.W.; Steven, A.C.; Wickner, R.B. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 1999, 283, 1339–1343. [Google Scholar] [CrossRef]
- Brachmann, A.; Baxa, U.; Wickner, R.B. Prion generation in vitro: Amyloid of Ure2p is infectious. EMBO J. 2005, 24, 3082–3092. [Google Scholar] [CrossRef]
- Cooper, T.G. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: Connecting the dots. FEMS Microbiol. Rev. 2002, 26, 223–238. [Google Scholar] [CrossRef]
- Cox, B.S. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 1965, 20, 505–521. [Google Scholar] [CrossRef]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996, 15, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Patino, M.M.; Liu, J.-J.; Glover, J.R.; Lindquist, S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996, 273, 622–626. [Google Scholar] [CrossRef]
- King, C.-Y.; Tittmann, P.; Gross, H.; Gebert, R.; Aebi, M.; Wuthrich, K. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 6618–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. In vitro propagation of the prion-like state of yeast Sup35 protein. Science 1997, 277, 381–383. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.R.; Kowal, A.S.; Shirmer, E.C.; Patino, M.M.; Liu, J.-J.; Lindquist, S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997, 89, 811–819. [Google Scholar] [CrossRef]
- King, C.-Y.; Diaz-Avalos, R. Protein-only transmission of three yeast prion strains. Nature 2004, 428, 319–323. [Google Scholar] [CrossRef]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef]
- Frolova, L.; LeGoff, X.; Rasmussen, H.H.; Cheperegin, S.; Drugeon, G.; Kress, M.; Arman, I.; Haenni, A.-L.; Celis, J.E.; Philippe, M.; et al. A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature 1994, 372, 701–703. [Google Scholar] [CrossRef]
- Stansfield, I.; Jones, K.M.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Poznyakovski, A.I.; Paushkin, S.V.; Nierras, C.R.; Cox, B.S.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [PubMed]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef]
- Sondheimer, N.; Lindquist, S. Rnq1: An epigenetic modifier of protein function in yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef] [PubMed]
- Daskalov, A.; Habenstein, B.; Martinez, D.; Debets, A.J.; Sabate, R.; Loquet, A.; Saupe, S.J. Signal transduction by a fungal NOD-like receptor based on propagation of a prion amyloid fold. PLoS Biol. 2015, 13, e1002059. [Google Scholar] [CrossRef]
- Cai, X.; Xu, H.; Chen, Z.J. Prion-like polymerization in immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2017, 9, a023580. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996, 144, 1375–1386. [Google Scholar]
- Schlumpberger, M.; Prusiner, S.B.; Herskowitz, I. Induction of distinct [URE3] yeast prion strains. Mol. Cell. Biol. 2001, 21, 7035–7046. [Google Scholar] [CrossRef]
- Bradley, M.E.; Edskes, H.K.; Hong, J.Y.; Wickner, R.B.; Liebman, S.W. Interactions among prions and prion “strains” in yeast. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16392–16399. [Google Scholar] [CrossRef]
- Hall, D.; Edskes, H.E. Silent prions lying in wait: A two-hit model of prion/amyloid formation and infection. J. Mol. Biol. 2004, 336, 775–786. [Google Scholar] [CrossRef]
- Kalastavadi, T.; True, H.L. Analysis of the [RNQ+] prion reveals stability of amyloid fibers as the key determinant of yeast prion variant propagation. J. Biol. Chem. 2010, 285, 20748–20755. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.; Kryndushkin, D.; Wickner, R.B. Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. USA 2011, 108, 5337–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edskes, H.K.; McCann, L.M.; Hebert, A.M.; Wickner, R.B. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics 2009, 181, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Bateman, D.A.; Wickner, R.B. [PSI+] prion transmission barriers protect Saccharomyces cerevisiae from infection: Intraspecies ‘species barriers’. Genetics 2012, 190, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Bateman, D.; Wickner, R.B. The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet. 2013, 9, e1003257. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, P.A.; Reidy, M.; Masison, D.C. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics 2011, 188, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Nguyen, P.P.; Patel, M.J.; Sporn, Z.A.; Hines, J.K. Functional diversification of Hsp40: Distinct J-protein functional requirements for two prions allow for chaperone-dependent prion selection. PLoS Genet. 2014, 10, e41004510. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Wickner, R.B. Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae. Mol. Biol. Cell. 2007, 18, 2149–2154. [Google Scholar] [CrossRef]
- Fan, Q.; Park, K.-W.; Du, Z.; Morano, K.A.; Li, L. The role of Sse1 in the de novo formation and variant determination of the [PSI+] prion. Genetcs 2007, 177, 1583–1593. [Google Scholar] [CrossRef]
- Gorkovskiy, A.; Reidy, M.; Masison, D.C.; Wickner, R.B. Hsp104 at normal levels cures many [PSI+] variants in a process promoted by Sti1p, Hsp90 and Sis1p. Proc. Natl. Acad. Sci. USA 2017, 114, E4193–E4202. [Google Scholar] [CrossRef]
- Wickner, R.B.; Beszonov, E.; Bateman, D.A. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc. Natl. Acad. Sci. USA 2014, 111, E2711–E2720. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Wickner, R.B. Nonsense-mediated mRNA decay factors cure most [PSI+] prion variants. Proc. Natl. Acad. Sci. USA 2018, 115, E1184–E1193. [Google Scholar] [CrossRef]
- Wickner, R.B.; Kelly, A.C.; Bezsonov, E.E.; Edskes, H.E. Prion propagation is controlled by inositol polyphosphates. Proc. Natl. Acad. Sci. USA 2017, 114, E8402–E8410. [Google Scholar] [CrossRef] [PubMed]
- Ter-Avanesyan, M.D.; Dagkesamanskaya, A.R.; Kushnirov, V.V.; Smirnov, V.N. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 1994, 137, 671–676. [Google Scholar] [PubMed]
- Masison, D.C.; Blanc, A.; Ribas, J.C.; Carroll, K.; Sonenberg, N.; Wickner, R.B. Decoying the cap- mRNA degradation system by a dsRNA virus and poly(A)- mRNA surveillance by a yeast antiviral system. Mol. Cell. Biol. 1995, 15, 2763–2771. [Google Scholar] [CrossRef] [PubMed]
- Masison, D.C.; Maddelein, M.-L.; Wickner, R.B. The prion model for [URE3] of yeast: Spontaneous generation and requirements for propagation. Proc. Natl. Acad. Sci. USA 1997, 94, 12503–12508. [Google Scholar] [CrossRef] [Green Version]
- Tycko, R. Molecular structure of amyloid fibrils: Insights from solid-state NMR. Quart. Rev. Biophys. 2006, 1, 1–55. [Google Scholar] [CrossRef]
- Tycko, R. Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. [Google Scholar] [CrossRef]
- Shewmaker, F.; Wickner, R.B.; Tycko, R. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc. Natl. Acad. Sci. USA 2006, 103, 19754–19759. [Google Scholar] [CrossRef]
- Baxa, U.; Wickner, R.B.; Steven, A.C.; Anderson, D.; Marekov, L.; Yau, W.-M.; Tycko, R. Characterization of β-sheet structure in Ure2p1-89 yeast prion fibrils by solid state nuclear magnetic resonance. Biochemistry 2007, 46, 13149–13162. [Google Scholar] [CrossRef]
- Wickner, R.B.; Dyda, F.; Tycko, R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure. Proc. Natl. Acad. Sci. USA 2008, 105, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Shewmaker, F.; Kryndushkin, D.; Chen, B.; Tycko, R.; Wickner, R.B. Two prion variants of Sup35p have in-register β-sheet structures, independent of hydration. Biochemistry 2009, 48, 5074–5082. [Google Scholar] [CrossRef] [PubMed]
- Gorkovskiy, A.; Thurber, K.R.; Tycko, R.; Wickner, R.B. Locating folds of the in-register parallel β-sheet of the Sup35p prion domain infectious amyloid. Proc. Natl. Acad. Sci. USA 2014, 111, E4615–E4622. [Google Scholar] [CrossRef] [PubMed]
- Ohhashi, Y.; Yamaguchi, Y.; Kurahashi, H.; Kamatari, Y.O.; Sugiyama, S.; Uluca, B.; Piechatzek, T.; Komi, Y.; Shida, T.; Muller, H.; et al. Molecular basis for diversification of yeast prion strain conformation. Proc. Natl. Acad. Sci. USA 2018, 115, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.; Maddelein, M.L.; Siemer, A.B.; Luhrs, T.; Ernst, M.; Meier, B.H.; Saupe, S.J.; Riek, R. Correlation of structural elements and infectivity of the HET-s prion. Nature 2005, 435, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s (218–279) prion form a beta solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Doel, S.M.; McCready, S.J.; Nierras, C.R.; Cox, B.S. The dominant PNM2− mutation which eliminates the [PSI] factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 1994, 137, 659–670. [Google Scholar] [PubMed]
- DePace, A.H.; Santoso, A.; Hillner, P.; Weissman, J.S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998, 93, 1241–1252. [Google Scholar] [CrossRef]
- Wickner, R.B.; Shewmaker, F.; Kryndushkin, D.; Edskes, H.K. Protein inheritance (prions) based on parallel in-register β-sheet amyloid structures. Bioessays 2008, 30, 955–964. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B.; Shewmaker, F.; Bateman, D.A.; Edskes, H.E.; Gorkovskiy, A.; Dayani, Y.; Bezsonov, E.E. Yeast prions: Structure, biology and prion-handling systems. Microbiol. Mol. Biol. Rev. 2015, 79, 1–17. [Google Scholar] [CrossRef]
- Pierce, M.M.; Baxa, U.; Steven, A.C.; Bax, A.; Wickner, R.B. Is the prion domain of soluble Ure2p unstructured? Biochemistry 2005, 44, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Kelly, M.J.; Gross, J.D.; Weissman, J.S. The structural basis of yeast prion strain variants. Nature 2007, 449, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Saupe, S.J. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Sem. Cell Dev. Biol. 2011, 22, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Eaglestone, S.S.; Cox, B.S.; Tuite, M.F. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J. 1999, 18, 1974–1981. [Google Scholar] [CrossRef] [PubMed]
- True, H.L.; Lindquist, S.L. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000, 407, 477–483. [Google Scholar] [CrossRef] [PubMed]
- True, H.L.; Berlin, I.; Lindquist, S.L. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 2004, 431, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Galopier, A.; Martini, C.; Matsufuji, S.; Fabret, C.; Rousset, C. Epigenetic control of polyamines by the prion [PSI+]. Nat. Cell. Biol. 2008, 10, 1069–1075. [Google Scholar] [CrossRef]
- Nakayashiki, T.; Kurtzman, C.P.; Edskes, H.K.; Wickner, R.B. Yeast prions [URE3] and [PSI+] are diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 10575–10580. [Google Scholar] [CrossRef]
- Kelly, A.C.; Shewmaker, F.P.; Kryndushkin, D.; Wickner, R.B. Sex, prions and plasmids in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, E2683–E2690. [Google Scholar] [CrossRef]
- Debets, A.J.; Dalstra, H.J.; Slakhorst, M.; Koopmanschap, B.; Hoekstra, R.F.; Saupe, S.J. High natural prevalence of a fungal prion. Proc. Natl. Acad. Sci. USA 2012, 109, 10432–10437. [Google Scholar] [CrossRef] [Green Version]
- Cuille, J.; Chelle, P.L. Experimental transmission of trembling to the goat. C. R. Seances Acad. Sci. 1939, 208, 1058–1060. [Google Scholar]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.-M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.-L.; Serban, D.; Carlson, G.A.; et al. Transgenic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Kochneva-Pervukhova, N.V.; Cechenova, M.B.; Frolova, N.S.; Ter-Avanesyan, M.D. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000, 19, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Santoso, A.; Chien, P.; Osherovich, L.Z.; Weissman, J.S. Molecular basis of a yeast prion species barrier. Cell 2000, 100, 277–288. [Google Scholar] [CrossRef]
- King, C.Y. Supporting the structural basis of prion strains: Induction and identification of [PSI] variants. J. Mol. Biol. 2001, 307, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Nakayashiki, T.; Ebihara, K.; Bannai, H.; Nakamura, Y. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol. Cell 2001, 7, 1121–1130. [Google Scholar] [CrossRef]
- Ter-Avanesyan, M.D.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Didichenko, S.A.; Chernoff, Y.O.; Inge-Vechtomov, S.G.; Smirnov, V.N. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 1993, 7, 683–692. [Google Scholar] [CrossRef]
- Mead, S.; Whitfield, J.; Poulter, M.; Shah, P.; Uphill, J.; Campbell, T.; Al-Dujaily, H.; Hummerch, H.; Beck, J.; Mein, C.A.; et al. A novel protective prion protein variant that colocalizes with Kuru exposure. N. Engl. J. Med. 2009, 361, 2056–2065. [Google Scholar] [CrossRef]
- Edskes, H.E.; Khamar, H.J.; Winchester, C.-L.; Greenler, A.J.; Zhou, A.; McGlinchey, R.P.; Gorkovskiy, A.; Wickner, R.B. Sporadic distribution of prion-forming ability of Sup35p from yeasts and fungi. Genetics 2014, 198, 605–616. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Cole, S.; Walker, C.A. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol. 1987, 68, 1875–1881. [Google Scholar] [CrossRef]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian evolution of prions in cell culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef]
- Collinge, J. Variant Creutzfeldt-Jakob disease. Lancet 1999, 354, 317–323. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef]
- Sharma, J.; Liebman, S.W. [PSI+] prion variant establishment in yeast. Mol. Microbiol. 2012, 86, 866–881. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Derkach, I.L.; Inge-Vechtomov, S.G. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr. Genet. 1993, 24, 268–270. [Google Scholar] [CrossRef]
- Zhou, P.; Derkatch, I.L.; Liebman, S.W. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI+] and [PIN+]. Mol. Microbiol. 2001, 39, 37–46. [Google Scholar] [CrossRef]
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73–82. [Google Scholar] [CrossRef]
- Schlieker, C.; Tews, I.; Bukau, B.; Mogk, A. Solubilization of aggregated proteins by ClpB/DnaK relies on the continuous extraction of unfolded polypeptides. FEBS Lett. 2004, 578, 351–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, R.; Tkach, J.M.; Vierling, E.; Glover, J.R. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem. 2004, 279, 29139–29146. [Google Scholar] [CrossRef] [PubMed]
- Lum, R.; Niggemann, M.; Glover, J.R. Peptide and protein binding in the axial channel of Hsp104. Insights into the mechanism of protein unfolding. J. Biol. Chem. 2008, 283, 30139–30146. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, R.D.; Bapat, K.; Newnam, G.P.; Zink, A.D.; Chernoff, Y.O. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol. Cell. Biol. 2001, 21, 4656–4669. [Google Scholar] [CrossRef] [PubMed]
- Ness, F.; Ferreira, P.; Cox, B.S.; Tuite, M.F. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol. Cell. Biol. 2002, 22, 5593–5605. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Jones, G.; Wegrzyn, R.D.; Masison, D.C. A role for cytosolic Hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics 2000, 156, 559–570. [Google Scholar]
- Jones, G.W.; Masison, D.C. Saccharomyces cerevisiae Hsp70 Mutations Affect [PSI(+)] Prion Propagation and Cell Growth Differently and Implicate Hsp40 and Tetratricopeptide Repeat Cochaperones in Impairment of [PSI(+)]. Genetics 2003, 163, 495–506. [Google Scholar]
- Roberts, B.T.; Moriyama, H.; Wickner, R.B. [URE3] prion propagation is abolished by a mutation of the primary cytosolic Hsp70 of budding yeast. Yeast 2004, 21, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Hines, J.K.; Li, X.; Du, Z.; Higurashi, T.; Li, L.; Craig, E.A. [SWI], the prion formed by the chromatin remodeling factor Swi1, is highly sensitive to alterations in Hsp70 chaperone system activity. PLoS Genet. 2011, 7, e1001309. [Google Scholar] [CrossRef]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Higurashi, T.; Hines, J.K.; Sahi, C.; Aron, R.; Craig, E.A. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc. Natl. Acad. Sci. USA 2008, 105, 16596–16601. [Google Scholar] [CrossRef] [Green Version]
- Troisi, E.M.; Rockman, M.E.; Nguyen, P.P.; Oliver, E.E.; Hines, J.K. Swa2, the yeast homolog of mammalian auxilin, is specifically required for the propagation of the prion variant [URE3-1]. Mol. Microbiol. 2015, 97, 926–941. [Google Scholar] [CrossRef]
- Reidy, M.; Miot, M.; Masison, D.C. Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics 2012, 192, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Gaur, D.; Gupta, A.; Puri, A.; Sharma, D. Hsp90-associated immunophilin homolog Cpr7 is required for the mitotic stability of [URE3] prion in Saccharomyces cerevisiae. PLoS Genet. 2015, 11, e1005567. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.-I.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Astor, M.T.; Kamiya, E.; Sporn, Z.A.; Berger, S.E.; Hines, J.K. Variant-specific and reciprocal Hsp40 functions in Hsp104-mediated prion elimination. Mol. Microbiol. 2018, 109, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, D.L.; Dobson, C.M.; Rachubinski, R.A. Chaperone proteins select and maintain [PIN+] prion conformations in Saccharomyces cerevisiae. J. Biol. Chem. 2013, 288, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Reidy, M.; Kumar, S.; Anderson, D.E.; Masison, D.C. Dual roles for yeast Sti1/Hop in regulating the Hsp90 chaperone cycle. Genetics 2018, 209, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Reidy, M.; Masison, D.C. Sti1 regulation of Hsp70 and Hsp90 is critical for curing of Saccharomyces cerevisiae [PSI+] prions by Hsp104. Mol. Cell. Biol. 2010, 30, 3542–3552. [Google Scholar] [CrossRef]
- Kiktev, D.A.; Patterson, J.C.; Muller, S.; Bariar, B.; Pan, T.; Chernoff, Y.O. Regulation of the chaperone effects on a yeast prion by the cochaperone Sgt2. Mol. Cell. Biol. 2012, 32, 4960–4970. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Shewmaker, F.; Wickner, R.B. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 2008, 27, 2725–2735. [Google Scholar] [CrossRef] [Green Version]
- Hung, G.C.; Masison, D.C. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 2006, 173, 611–620. [Google Scholar] [CrossRef]
- Helsen, C.W.; Glover, J.R. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104). J. Biol. Chem. 2012, 287, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.N.; Zhou, X.; Yim, Y.I.; Todor, H.; Ellerbrock, R.; Reidy, M.; Eisenberg, E.; Masison, D.C.; Greene, L.E. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot. Cell 2014, 13, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Ness, F.; Cox, B.; Wonwigkam, J.; Naeimi, W.R.; Tuite, M.F. Over-expression of the molecular chaperone Hsp104 in Saccharomyces cerevisiae results in the malpartition of [PSI+] propagons. Mol. Microbiol. 2017, 104, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, B.; Wongwigkam, J.; Tuite, M.F. Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI+] prion but not for prion propagation. Yeast 2010, 27, 167–179. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Smirnov, V.N.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Increased expression of Hsp40 chaperones, transcriptional factors, and ribosomal protein Rpp0 can cure yeast prions. J. Biol. Chem. 2002, 277, 23702–23708. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, H.; Edskes, H.K.; Wickner, R.B. [URE3] prion propagation in Saccharomyces cerevisiae: Requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol. Cell. Biol. 2000, 20, 8916–8922. [Google Scholar] [CrossRef]
- Kiktev, D.A.; Melomed, M.M.; Lu, C.D.; Newnam, G.P.; Chernoff, Y.O. Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol. Microbiol. 2015, 96, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Harashima, T.; Heitman, J. The Galpha protein Gpa2 controls yeast differentiation by interacting with kelch repeat proteins that mimic Gbeta subunits. Mol. Cell 2002, 10, 163–173. [Google Scholar] [CrossRef]
- Ishiwata, M.; Kurahashi, H.; Nakamura, Y. A G-protein γ subunit mimic is a general antagonist of prion propagation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2009, 106, 791–796. [Google Scholar] [CrossRef]
- Wickner, R.B.; Bezsonov, E.E.; Son, M.; Ducatez, M.; DeWilde, M.; Edskes, H.E. Anti-prion systems in yeast and inositol polyphosphates. Biochemistry 2018, 57, 1285–1292. [Google Scholar] [CrossRef]
- Koplin, A.; Preissler, S.; Ilina, Y.; Kock, M.; Scior, A.; Erhardt, M.; Deuerling, E. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J. Cell Biol. 2010, 189, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernoff, Y.O.; Newnam, G.P.; Kumar, J.; Allen, K.; Zink, A.D. Evidence for a protein mutator in yeast: Role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI+] prion. Mol. Cell. Biol. 1999, 19, 8103–8112. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Szczesniak, B.; Kochneva-Pervukhova, N.V.; Kushnirov, V.V.; Ter-Avanesyan, M.D.; Boguta, M. Ssb1 chaperone is a [PSI+] prion-curing factor. Curr. Genet. 2001, 39, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Ihrke, G.; Piermartiri, T.C.; Shewmaker, F. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 2012, 86, 1531–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kama, R.; Robinson, M.; Gerst, J.E. Btn2, a Hook1 ortholog and potential Batten disease-related protein, mediates late endosome-Golgi protein sorting in yeast. Mol. Cell. Biol. 2007, 27, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Steidle, E.A.; Chong, L.S.; Wu, M.; Crooke, E.; Fiedler, D.; Resnick, A.C.; Rolfes, R.J. A novel inositol pyrophosphate phosphatase in Saccharomyces cerevisiae: Siw14 protein selectively cleaves the β-phosphate from 5-diphosphoinositol pentakisphosphate (5PP-IP5). J. Biol. Chem. 2016, 291, 6772–6783. [Google Scholar] [CrossRef] [PubMed]
- Stein, K.C.; True, H.L. Structural variants of yeast prions show conformer-specific requirements for chaperone activity. Mol. Microbiol. 2014, 93, 1156–1171. [Google Scholar] [CrossRef]
- Sporn, M.A.; Hines, J.K. Hsp40 function in yeast prion propagation: Amyloid diversity necessitates chaperone functional complexity. Prion 2005, 9, 80–89. [Google Scholar] [CrossRef]
- Sadish, H.; Rampelt, H.; Shorter, J.; Wegrzyn, R.D.; Andreasson, C.; Lindquist, S.; Bukau, B. Hsp110 chaperones regulate prion formation and propagation in S. cerevisiae by two discrete activities. PLoS ONE 2008, 3, e1763. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant-Defining Condition/Trait | Prions Affected | Mechanism | Relation to Strong/Weak? | Refs. |
|---|---|---|---|---|
| Strength of phenotype (strong/weak) | [PSI+], [URE3] | high filament number adsorb more prion protein | [10,28,29] | |
| Prion stability | [PSI+], [URE3] | high filament number insures both daughters infected | Strong often stable, weak often unstable; exceptions | [10,28,29] |
| prion toxicity (all variants detrimental but the degree varies) | [PSI+], [URE3] | [PSI+]: depletion of Sup35p (essential) [URE3]: toxic effect of amyloid form | unknown | [33] |
| interspecies or intraspecies barriers | [PSI+], [URE3] | inefficient binding to amyloid of different protein sequence | [34,35,36] | |
| lethality or prion loss in Sis1p partial deletions | [PSI+] | unknown | strong variant lethal; weak variant lost | [37,38] |
| Sse1p required for propagation or generation | [PSI+] | strong variant weakened, weak variant lost | [39,40] | |
| curing by normal levels of Hsp104 | [PSI+] | some of both are cured; no relation to seed # | [41] | |
| curing by normal levels of Btn2p | [URE3] | filaments sequestered by Btn1p, Hsp42p | All cured variants are weak, but some weak variants not cured | [42] |
| curing by normal levels of Cur1p | [URE3] | unknown | All cured variants are weak, but some weak variants not cured | [42] |
| curing by normal levels of Upf1,2,3 | [PSI+] | complex formation with Sup35p | no relation to strong/weak | [43] |
| curing by normal levels of Siw14p | [PSI+] | limits 5PP-IP5 levels | unknown | [44] |
| Chaperone | Effects | Prions Affected | Refs |
|---|---|---|---|
| Hsp104 | filament cleavage (with Hsp70 and Hsp40) | all amyloid-based yeast prions | [104] |
| Ssa1–4 (Hsp70) | filament cleavage | [PSI+] and [URE3] | [96] |
| Sis1 (Hsp40) | propagation; needed for Hsp104 curing; prevents toxicity | all amyloid-based yeast prions; [PSI+]; strong [PSI+] | [37,100] |
| Swa2 (Hsp40) | propagation | [URE3] | [101] |
| Apj1 (Hsp40) | needed for Hsp104 curing of strong [PSI+] | [PSI+] | [105] |
| Hsp90 | needed for Hsp104 curing; variant selection; propagation | [PSI+], [PIN+], and [URE3] | [101,103,106,107] |
| Sti1 | needed for Hsp104 curing | [PSI+] | [108] |
| Cpr7 | Hsp90 co-chaperone | [URE3] | [103] |
| Fes1 | overproduction curing | [URE3] | [39] |
| Sse1 | propagation: necessary and overproduction curing | [URE3] and [PSI+] | [39,40] |
| Sgt2 | affects Hsp104 curing; induced by prions | [PSI+] and [PIN+] | [109] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wickner, R.B.; Son, M.; Edskes, H.K. Prion Variants of Yeast are Numerous, Mutable, and Segregate on Growth, Affecting Prion Pathogenesis, Transmission Barriers, and Sensitivity to Anti-Prion Systems. Viruses 2019, 11, 238. https://0-doi-org.brum.beds.ac.uk/10.3390/v11030238
Wickner RB, Son M, Edskes HK. Prion Variants of Yeast are Numerous, Mutable, and Segregate on Growth, Affecting Prion Pathogenesis, Transmission Barriers, and Sensitivity to Anti-Prion Systems. Viruses. 2019; 11(3):238. https://0-doi-org.brum.beds.ac.uk/10.3390/v11030238
Chicago/Turabian StyleWickner, Reed B., Moonil Son, and Herman K. Edskes. 2019. "Prion Variants of Yeast are Numerous, Mutable, and Segregate on Growth, Affecting Prion Pathogenesis, Transmission Barriers, and Sensitivity to Anti-Prion Systems" Viruses 11, no. 3: 238. https://0-doi-org.brum.beds.ac.uk/10.3390/v11030238