Evolutionary Dynamics of the Lineage 2 West Nile Virus That Caused the Largest European Epidemic: Italy 2011–2018

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Samples and Datasets
2.3. Study of Recombinations and Phylogenetic Analysis
2.4. Models for Estimating Distances
2.5. Selective Pressure
2.6. Phylogeographical Analysis
2.7. Bayesian Coalescent and Birth-Death Skyline Analyses
3. Results
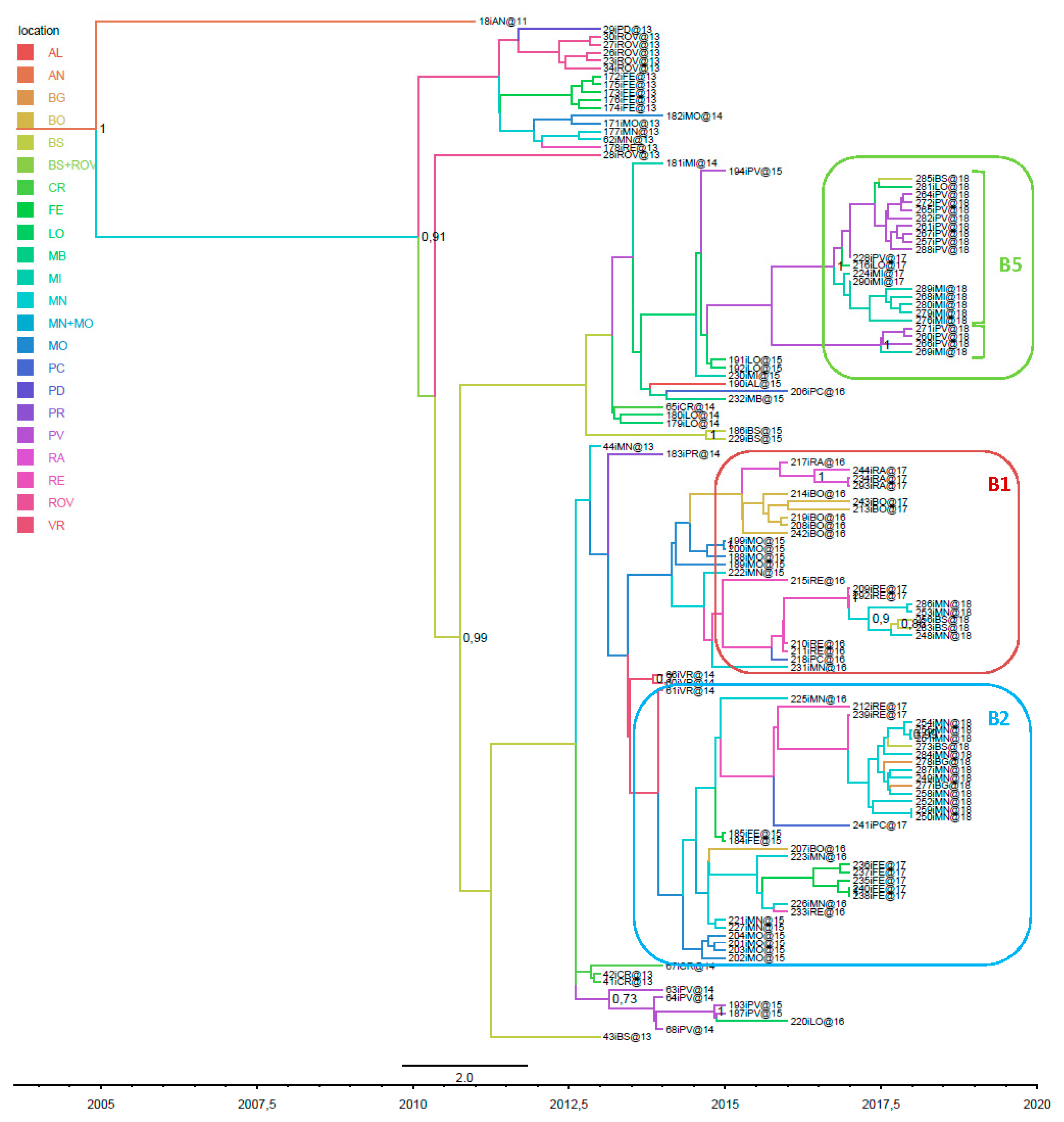
3.1. Phylogenetic Analysis of Italian Isolates
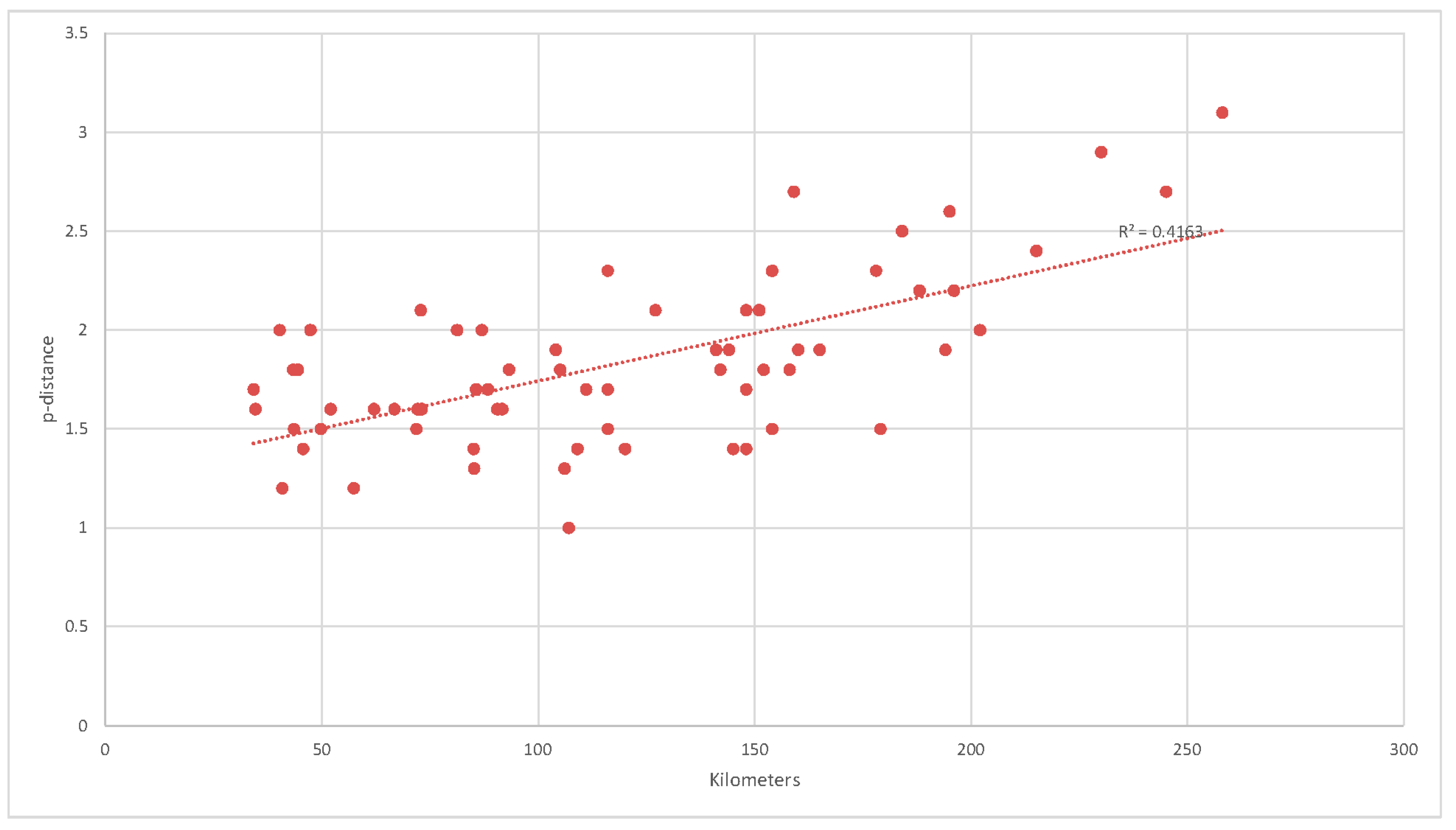
3.2. Genetic Distances
3.3. Differences in Amino Acids
3.4. Phylogeographical Analysis of the European and Italian WNV-2 Clades
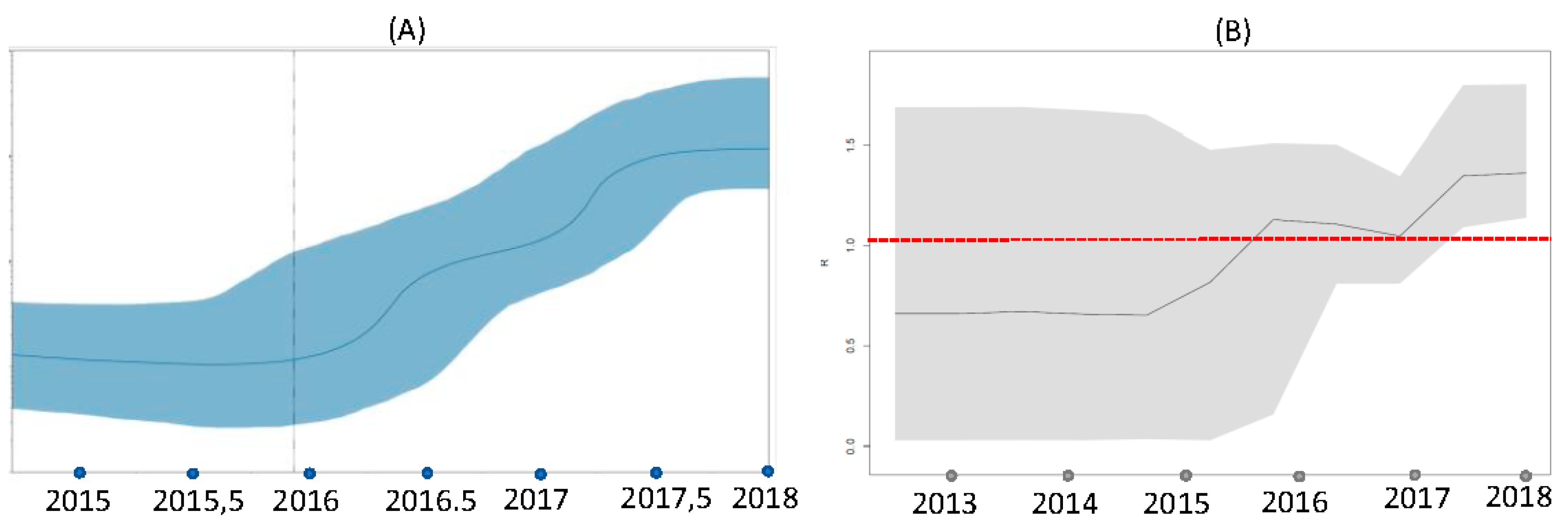
3.5. Phylodynamics (Coalescent and Birth-Death Analyses)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rizzo, C.; Napoli, C.; Venturi, G.; Pupella, S.; Lombardini, L.; Calistri, P.; Monaco, F.; Cagarelli, R.; Angelini, P.; Bellini, R.; et al. West Nile virus transmission: Results from the integrated surveillance system in Italy, 2008 to 2015. Euro. Surveill. 2016, 21, 30340. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Ebel, G.D.; Deubel, V.; Kerst, A.J.; Murri, S.; Meyer, R.; Bowen, M.; McKinney, N.; Morrill, W.E.; Crabtree, M.B.; et al. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology 2002, 298, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Ferenczi, E.; Erdelyi, K.; Kutasi, O.; Csorgo, T.; Seidel, B.; Weissenbock, H.; Brugger, K.; Ban, E.; Nowotny, N. Explosive spread of a neuroinvasive lineage 2 West Nile virus in Central Europe, 2008/2009. Vet. Microbiol. 2013, 165, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Ivanics, E.; Erdelyi, K.; Ursu, K.; Ferenczi, E.; Weissenbock, H.; Nowotny, N. Lineage 1 and 2 strains of encephalitic West Nile virus, central Europe. Emerg. Infect. Dis. 2006, 12, 618–623. [Google Scholar] [CrossRef]
- Danis, K.; Papa, A.; Theocharopoulos, G.; Dougas, G.; Athanasiou, M.; Detsis, M.; Baka, A.; Lytras, T.; Mellou, K.; Bonovas, S.; et al. Outbreak of West Nile virus infection in Greece, 2010. Emerg. Infect. Dis. 2011, 17, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Platonov, A.E.; Karan, L.S.; Shopenskaia, T.A.; Fedorova, M.V.; Koliasnikova, N.M.; Rusakova, N.M.; Shishkina, L.V.; Arshba, T.E.; Zhuravlev, V.I.; Govorukhina, M.V.; et al. Genotyping of West Nile fever virus strains circulating in southern Russia as an epidemiological investigation method: Principles and results. Zh. Mikrobiol. Epidemiol. Immunobiol. 2011, 2, 29–37. [Google Scholar]
- Autorino, G.L.; Battisti, A.; Deubel, V.; Ferrari, G.; Forletta, R.; Giovannini, A.; Lelli, R.; Murri, S.; Scicluna, M.T. West Nile virus epidemic in horses, Tuscany region, Italy. Emerg. Infect. Dis. 2002, 8, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Calistri, P.; Giovannini, A.; Savini, G.; Monaco, F.; Bonfanti, L.; Ceolin, C.; Terregino, C.; Tamba, M.; Cordioli, P.; Lelli, R. West Nile virus transmission in 2008 in north-eastern Italy. Zoonoses Public Health 2010, 57, 211–219. [Google Scholar] [CrossRef]
- Rossini, G.; Cavrini, F.; Pierro, A.; Macini, P.; Finarelli, A.; Po, C.; Peroni, G.; Di Caro, A.; Capobianchi, M.; Nicoletti, L.; et al. First human case of West Nile virus neuroinvasive infection in Italy, September 2008—Case report. Euro. Surveill. 2008, 13, 19002. [Google Scholar] [CrossRef]
- Rizzo, C.; Vescio, F.; Declich, S.; Finarelli, A.C.; Macini, P.; Mattivi, A.; Rossini, G.; Piovesan, C.; Barzon, L.; Palu, G.; et al. West Nile virus transmission with human cases in Italy, August–September 2009. Euro. Surveill. 2009, 14, 19353. [Google Scholar]
- Magurano, F.; Remoli, M.E.; Baggieri, M.; Fortuna, C.; Marchi, A.; Fiorentini, C.; Bucci, P.; Benedetti, E.; Ciufolini, M.G.; Rizzo, C.; et al. Circulation of West Nile virus lineage 1 and 2 during an outbreak in Italy. Clin. Microbiol. Infect. 2012, 18, E545–E547. [Google Scholar] [CrossRef] [Green Version]
- Riccardo, F.; Monaco, F.; Bella, A.; Savini, G.; Russo, F.; Cagarelli, R.; Dottori, M.; Rizzo, C.; Venturi, G.; Di Luca, M.; et al. An early start of West Nile virus seasonal transmission: The added value of One Heath surveillance in detecting early circulation and triggering timely response in Italy, June to July 2018. Euro. Surveill. 2018, 23, 1800427. [Google Scholar] [CrossRef] [PubMed]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genomics 2008, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus. Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Williamson, C.; Posada, D. RDP2: Recombination detection and analysis from sequence alignments. Bioinformatics 2005, 21, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. Journal of molecular evolution 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS. Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Heled, J.; Drummond, A.J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Stadler, T.; Kuhnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl. Acad. Sci. USA 2013, 110, 228–233. [Google Scholar] [CrossRef]
- Zehender, G.; Veo, C.; Ebranati, E.; Carta, V.; Rovida, F.; Percivalle, E.; Moreno, A.; Lelli, D.; Calzolari, M.; Lavazza, A.; et al. Reconstructing the recent West Nile virus lineage 2 epidemic in Europe and Italy using discrete and continuous phylogeography. PLoS ONE 2017, 12, e0179679. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Salcuni, P.; Nicoletti, L.; Ciufolini, M.G.; Russo, F.; Masala, R.; Frongia, O.; Finarelli, A.C.; Gramegna, M.; Gallo, L.; et al. Epidemiological surveillance of West Nile neuroinvasive diseases in Italy, 2008 to 2011. Euro. Surveill. 2012, 17, 20172. [Google Scholar] [PubMed]
- Calzolari, M.; Pautasso, A.; Montarsi, F.; Albieri, A.; Bellini, R.; Bonilauri, P.; Defilippo, F.; Lelli, D.; Moreno, A.; Chiari, M.; et al. West Nile Virus Surveillance in 2013 via Mosquito Screening in Northern Italy and the Influence of Weather on Virus Circulation. PLoS ONE 2015, 10, e0140915. [Google Scholar] [CrossRef] [PubMed]
- Savini, G.; Capelli, G.; Monaco, F.; Polci, A.; Russo, F.; Di Gennaro, A.; Marini, V.; Teodori, L.; Montarsi, F.; Pinoni, C.; et al. Evidence of West Nile virus lineage 2 circulation in Northern Italy. Vet. Microbiol. 2012, 158, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Bagnarelli, P.; Marinelli, K.; Trotta, D.; Monachetti, A.; Tavio, M.; Del Gobbo, R.; Capobianchi, M.; Menzo, S.; Nicoletti, L.; Magurano, F.; et al. Human case of autochthonous West Nile virus lineage 2 infection in Italy, September 2011. Euro. Surveill. 2011, 16, 20002. [Google Scholar] [PubMed]
- Armstrong, P.M.; Vossbrinck, C.R.; Andreadis, T.G.; Anderson, J.F.; Pesko, K.N.; Newman, R.M.; Lennon, N.J.; Birren, B.W.; Ebel, G.D.; Henn, M.R. Molecular evolution of West Nile virus in a northern temperate region: Connecticut, USA 1999–2008. Virology 2011, 417, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Lefkowitz, E.J. Recombination in West Nile Virus: Minimal contribution to genomic diversity. Virol J. 2009, 6, 165. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Weger-Lucarelli, J.; Murrieta, R.A.; Fauver, J.R.; Garcia-Luna, S.M.; Prasad, A.N.; Black, W.C.t.; Ebel, G.D. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host Switching. Cell Host Microbe. 2016, 19, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, F.; Savini, G.; Calistri, P.; Polci, A.; Pinoni, C.; Bruno, R.; Lelli, R. 2009 West Nile disease epidemic in Italy: First evidence of overwintering in Western Europe? Res. Vet. Sci. 2011, 91, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, I.; Betasova, L.; Blazejova, H.; Venclikova, K.; Strakova, P.; Sebesta, O.; Mendel, J.; Bakonyi, T.; Schaffner, F.; Nowotny, N.; et al. West Nile virus in overwintering mosquitoes, central Europe. Parasit. Vectors 2017, 10, 452. [Google Scholar] [CrossRef]
- Montecino-Latorre, D.; Barker, C.M. Overwintering of West Nile virus in a bird community with a communal crow roost. Sci. Rep. 2018, 8, 6088. [Google Scholar]
- Woolhouse, M.E.; Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 2005, 11, 1842–1847. [Google Scholar] [CrossRef]
- Kilpatrick, A.M.; Randolph, S.E. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet 2012, 380, 1946–1955. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WNV Group | 2013 | 2014 | 2015 | 2016 | 2017 | 2018 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N1 | L2 | N | L | N | L | N | L | N | L | N | L | |
| A (17) | 16 | VE3 LO4 E5 | 1 | E | - | - | - | - | - | - | - | - |
| B (49) | ||||||||||||
| B1 (15) | 1 | LO | 1 | E | 4 | E | 6 | LO-E | 1 | E | 2 | LO |
| B2 (15) | - | - | 1 | VE | 6 | E | 4 | LO-E | 2 | E | 2 | LO |
| B3 (3) | - | - | 3 | LO | - | - | - | - | - | - | - | - |
| B4 (5) | - | - | - | - | 4 | LO | 1 | LO | - | - | - | - |
| B5 (8) | - | - | 1 | LO | 1 | LO | - | - | 1 | LO | 5 | LO |
| B6 (3) | - | - | - | - | 2 | LO | 1 | LO | - | - | - | - |
| Within | A | B | Total |
|---|---|---|---|
| Mean p distance (SE) | 1.08 (0.16) | 1.84 (0.22) | 1.73 (0.18) |
| n° of differences (SE) | 9.67 (1.31) | 16.49 (1.62) | 15.47 (1.62) |
| Synonymous (SE) | 3.16 (0.65) | 5.93 (0.76) | 5.44 (0.71) |
| Non synonymous (SE) | 0.42 (0.10) | 0.48 (0.12) | 0.51 (0.1) |
| Between | |||
| Mean p-distance (SE) | - | 1.71 (0.18) | - |
| n° of differences (SE) | - | 15,32 (1.89) | - |
| Synonymous (SE) | - | 5.36 (0.83) | - |
| Non synonymous (SE) | - | 0.52 (0.12) |
| Within | |
| Years | Mean P-Distance (SE) |
| 2013 | 0.55 (0.18) |
| 2014 | 1.1 (0.12) |
| 2015 | 1.49 (0.19) |
| 2016 | 1.61 (0.22) |
| 2017 | 2.38 (0.33) |
| 2018 | 2.54 (0.25) |
| Total | 1.67 (0.18) |
| Between | |
| Δ Years | Mean P-Distance (SE) |
| 1 | 1.64 (0.56) |
| 2 | 1.77 (0.5) |
| 3 | 1.83 (0.48) |
| 4 | 1.94 (0.27) |
| 5 | 1.94 (0.27) |
| NO OF VARIANTS (/76) | % | AA CHANGES | POSITION | GENE | METHOD |
|---|---|---|---|---|---|
| 75 | 0.99 | V -> A | 416 (139) | prM | - |
| 17 | 0.23 | T -> A | 427 (143) | prM | - |
| 3 | 0.04 | I -> S | 473 (158) | prM | - |
| 15 | 0.2 | A -> T | 1021 (341) | ENV | - |
| 17 | 0.23 | H -> Y | 2476 (826) | NS1 | - |
| 5 | 0.07 | I -> V | 2509 (837) | NS1 | - |
| 4 | 0.05 | I -> F | 2740 (914) | NS1 | - |
| 17 | 0.23 | A -> V | 2810 (937) | NS1 | - |
| 75 | 0.99 | V -> A | 2933 (978) | NS1 | - |
| 75 | 0.99 | S -> C | 4003 (1335) | NS2A | - |
| 3 | 0.04 | R -> G | 5263 (1755) | NS3 | - |
| 14 | 0.18 | I -> V | 5863 (1955) | NS3 | - |
| 75 | 0.99 | T -> I | 6629 (2210) | NS4A | - |
| 6 | 0.08 | H -> R | 7712 (2571) | NS5 | IFEL |
| 17 | 0.23 | Y -> H | 8191 (2731) | NS5 | IFEL |
| 75 | 0.99 | S -> G | 8266 (2756) | NS5 | - |
| 18 | 0.24 | N -> S | 8603 (2868) | NS5 | IFEL |
| 75 | 0.99 | H -> R | 8924 (2975) | NS5 | - |
| 75 | 0.99 | A -> T | 9535 (3179) | NS5 | - |
| 18 | 0.24 | V -> A | 10241 (3414) | NS5 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veo, C.; della Ventura, C.; Moreno, A.; Rovida, F.; Percivalle, E.; Canziani, S.; Torri, D.; Calzolari, M.; Baldanti, F.; Galli, M.; et al. Evolutionary Dynamics of the Lineage 2 West Nile Virus That Caused the Largest European Epidemic: Italy 2011–2018. Viruses 2019, 11, 814. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090814
Veo C, della Ventura C, Moreno A, Rovida F, Percivalle E, Canziani S, Torri D, Calzolari M, Baldanti F, Galli M, et al. Evolutionary Dynamics of the Lineage 2 West Nile Virus That Caused the Largest European Epidemic: Italy 2011–2018. Viruses. 2019; 11(9):814. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090814
Chicago/Turabian StyleVeo, Carla, Carla della Ventura, Ana Moreno, Francesca Rovida, Elena Percivalle, Sabrina Canziani, Debora Torri, Mattia Calzolari, Fausto Baldanti, Massimo Galli, and et al. 2019. "Evolutionary Dynamics of the Lineage 2 West Nile Virus That Caused the Largest European Epidemic: Italy 2011–2018" Viruses 11, no. 9: 814. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090814