HIV-1 Unique Recombinant Forms Identified in Slovenia and Their Characterization by Near Full-Length Genome Sequencing


Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Subtyping of Pol Region
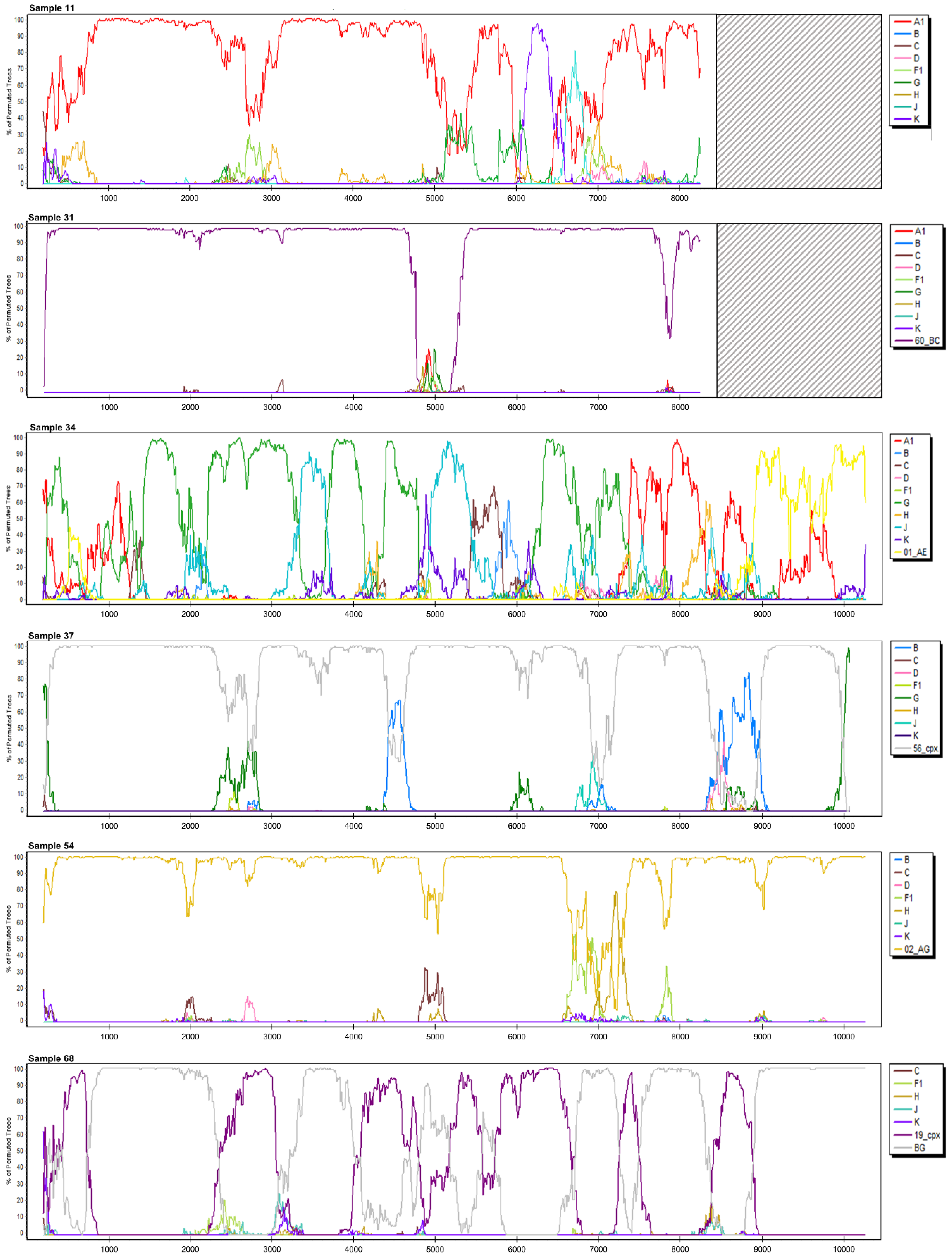
3.2. Near Full-Length HIV Genome Sequencing and Characterization
3.2.1. Sample 11
3.2.2. Sample 12
3.2.3. Sample 25
3.2.4. Sample 29
3.2.5. Sample 31
3.2.6. Sample 34
3.2.7. Sample 37
3.2.8. Sample 39
3.2.9. Sample 43
3.2.10. Sample 54
3.2.11. Sample 56
3.2.12. Sample 65
3.2.13. Sample 68
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- UNAIDS. Global HIV & AIDS Statistics—2019 Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 16 October 2019).
- Ghosn, J.; Taiwo, B.; Seedat, S.; Autran, B.; Katlama, C. HIV. Lancet 2018, 392, 685–697. [Google Scholar] [CrossRef]
- HIV Circulating Recombinant Forms (CRFs). Available online: https://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html (accessed on 18 October 2019).
- Robertson, D.L.; Anderson, J.P.; Bradac, J.A.; Carr, J.K.; Foley, B.; Funkhouser, R.K.; Gao, F.; Hahn, B.H.; Kalish, M.L.; Kuiken, C.; et al. HIV-1 nomenclature proposal. Science 2000, 288, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; McArthur, C.; Vallari, A.; Sthreshley, L.; Cloherty, G.A.; Berg, M.G.; Rodgers, M.A. Complete genome sequence of CG-0018a-01 establishes HIV-1 subtype L. J. Acquir. Immune Defic. Syndr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Fleminger, I.; Kirtley, S.; Williams, B.; Gouws-Williams, E.; Ghys, P.D.; WHO–UNAIDS Network for HIV Isolation Characterisation. Global and regional molecular epidemiology of HIV-1, 1990–2015: A systematic review, global survey, and trend analysis. Lancet Infect. Dis. 2019, 19, 143–155. [Google Scholar] [CrossRef]
- Babič, D.Z.; Zelnikar, M.; Seme, K.; Vandamme, A.M.; Snoeck, J.; Tomažič, J.; Vidmar, L.; Karner, P.; Poljak, M. Prevalence of antiretroviral drug resistance mutations and HIV-1 non-B subtypes in newly diagnosed drug-naïve patients in Slovenia, 2000–2004. Virus Res. 2006, 118, 156–163. [Google Scholar] [CrossRef]
- Lunar, M.M.; Židovec Lepej, S.; Abecasis, A.B.; Tomažič, J.; Vidmar, L.; Karner, P.; Vovko, T.D.; Pečavar, B.; Maver, P.J.; Seme, K.; et al. Short communication: Prevalence of HIV type 1 transmitted drug resistance in Slovenia: 2005–2010. AIDS Res. Hum. Retrovir. 2013, 29, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Lunar, M.M.; Židovec Lepej, S.; Tomažič, J.; Vovko, T.D.; Pečavar, B.; Turel, G.; Maver, M.; Poljak, M. HIV-1 transmitted drug resistance in Slovenia and its impact on predicted treatment effectiveness: 2011–2016 update. PLoS ONE 2018, 13, e0196670. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, T.; Deforche, K.; Cassol, S.; Salminen, M.; Paraskevis, D.; Seebregts, C.; Snoeck, J.; van Rensburg, E.J.; Wensing, A.M.; van de Vijver, D.A.; et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics 2005, 21, 3797–3800. [Google Scholar] [CrossRef] [Green Version]
- Pineda-Peña, A.C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gómez-López, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Schmit, J.C.; Bercoff, D.P. COMET: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.K.; Zhang, M.; Bulla, I.; Leitner, T.; Korber, B.; Morgenstern, B.; Stanke, M. jpHMM: Improving the reliability of recombination prediction in HIV-1. Nucleic Acids Res. 2009, 37, W647–W651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Posada, D.; Stawiski, E.; Chappey, C.; Poon, A.F.; Hughes, G.; Fearnhill, E.; Gravenor, M.B.; Leigh Brown, A.J.; Frost, S.D. An evolutionary model-based algorithm for accurate phylogenetic breakpoint mapping and subtype prediction in HIV-1. PLoS Comput. Biol. 2009, 5, e1000581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerenwinkel, N.; Däumer, M.; Oette, M.; Korn, K.; Hoffmann, D.; Kaiser, R.; Lengauer, T.; Selbig, J.; Walter, H. Geno2pheno: Estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic Acids Res. 2003, 31, 3850–3855. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [Green Version]
- HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html (accessed on 23 February 2018).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 11 December 2018).
- Gall, A.; Ferns, B.; Morris, C.; Watson, S.; Cotton, M.; Robinson, M.; Berry, N.; Pillay, D.; Kellam, P. Universal amplification, next-generation sequencing, and assembly of HIV-1 genomes. J. Clin. Microbiol. 2012, 50, 3838–3844. [Google Scholar] [CrossRef] [Green Version]
- Wymant, C.; Blanquart, F.; Golubchik, T.; Gall, A.; Bakker, M.; Bezemer, D.; Croucher, N.J.; Hall, M.; Hillebregt, M.; Ong, S.H.; et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol. 2018, 4, vey007. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.; Gall, A.; Ong, S.H.; Brener, J.; Ferns, B.; Goulder, P.; Nastouli, E.; Keane, J.A.; Kellam, P.; Otto, T.D. IVA: Accurate de novo assembly of RNA virus genomes. Bioinformatics 2015, 31, 2374–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Fastaq. Available online: https://github.com/sanger-pathogens/Fastaq (accessed on 8 May 2019).
- SMALT. Available online: https://www.sanger.ac.uk/science/tools/smalt-0 (accessed on 8 May 2019).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recombinant HIV-1 Drawing Tool. Available online: https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html (accessed on 3 September 2019).
- Lai, A.; Simonetti, F.R.; Brindicci, G.; Bergna, A.; Di Giambenedetto, S.; Sterrantino, G.; Mussini, C.; Menzo, S.; Bagnarelli, P.; Zazzi, M.; et al. Local epidemics gone viral: Evolution and diffusion of the Italian HIV-1 recombinant form CRF60_BC. Front. Microbiol. 2019, 10, 769. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.; Lebedeva, N.; Moskaleychik, F.; Pronin, A.; Kazennova, E.; Bobkova, M. Human immunodeficiency virus-1 diversity in the Moscow region, Russia: Phylodynamics of the most common subtypes. Front. Microbiol. 2019, 10, 320. [Google Scholar] [CrossRef]
- Díez-Fuertes, F.; Cabello, M.; Thomson, M.M. Bayesian phylogeographic analyses clarify the origin of the HIV-1 subtype A variant circulating in former Soviet Union’s countries. Infect. Genet. Evol. 2015, 33, 197–205. [Google Scholar] [CrossRef]
- Machado, L.Y.; Blanco, M.; López, L.S.; Díaz, H.M.; Dubed, M.; Valdés, N.; Noa, E.; Martínez, L.; Pérez, M.T.; Romay, D.M.; et al. National survey of pre-treatment HIV drug resistance in Cuban patients. PLoS ONE 2019, 14, e0221879. [Google Scholar] [CrossRef]
- Galindo, J.A.P.; Torres-Puente, M.; Gimeno, C.; Ortega, E.; Navarro, D.; Galindo, M.J.; Navarro, L.; Navarro, V.; Juan, A.; Belda, J.; et al. Expansion of the CRF19_cpx variant in Spain. J. Clin. Virol. 2015, 69, 146–149. [Google Scholar] [CrossRef]
- González-Domenech, C.M.; Viciana, I.; Delaye, L.; Mayorga, M.L.; Palacios, R.; de la Torre, J.; Jarilla, F.; Castaño, M.; Del Arco, A.; Clavijo, E.; et al. Emergence as an outbreak of the HIV-1 CRF19_cpx variant in treatment-naïve patients in southern Spain. PLoS ONE 2018, 13, e0190544. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Parra, S.; Álvarez, M.; Fernandez-Caballero, J.A.; Pérez, A.B.; Santos, J.; Bisbal, O.; Aguilera, A.; Rivero, M.; García-Fraile, L.; García, F. Continued propagation of the CRF19_cpx variant among HIV-positive MSM patients in Spain. J. Antimicrob. Chemother. 2018, 73, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Fernández-García, A.; Delgado, E.; Cuevas, M.T.; Vega, Y.; Montero, V.; Sánchez, M.; Carrera, C.; López-Álvarez, M.J.; Miralles, C.; Pérez-Castro, S.; et al. Identification of an HIV-1 BG intersubtype recombinant form (CRF73_BG), partially related to CRF14_BG, which is circulating in Portugal and Spain. PLoS ONE 2016, 11, e0148549. [Google Scholar] [CrossRef] [PubMed]
- Beamud, B.; Bracho, M.A.; González-Candelas, F. Characterization of new recombinant forms of HIV-1 from the Comunitat Valenciana (Spain) by phylogenetic incongruence. Front. Microbiol. 2019, 10, 1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouri, V.; Khouri, R.; Alemán, Y.; Abrahantes, Y.; Vercauteren, J.; Pineda-Peña, A.C.; Theys, K.; Megens, S.; Moutschen, M.; Pfeifer, N.; et al. CRF19_cpx is an evolutionary fit HIV-1 variant strongly associated with rapid progression to AIDS in Cuba. EBioMedicine 2015, 2, 244–254. [Google Scholar] [CrossRef] [Green Version]
- EACS Guidelines Version 8.1, October 2016. Available online: https://www.eacsociety.org/files/guidelines_8.1-english.pdf (accessed on 5 November 2019).
- Delatorre, E.; Bello, G. Time-scale of minor HIV-1 complex circulating recombinant forms from Central and West Africa. BMC Evol. Biol. 2016, 16, 249. [Google Scholar] [CrossRef] [Green Version]
- Leoz, M.; Feyertag, F.; Charpentier, C.; Delaugerre, C.; Wirden, M.; Lemee, V.; Plantier, J.C. Characterization of CRF56_cpx, a new circulating B/CRF02/G recombinant form identified in MSM in France. AIDS 2013, 27, 2309–2312. [Google Scholar] [CrossRef]
- Tariq, U.; Parveen, A.; Akhtar, F.; Mahmood, F.; Ali, S.; Abidi, S.H. Emergence of circulating recombinant form 56_cpx in Pakistan. AIDS Res. Hum. Retrovir. 2018, 34, 1002–1004. [Google Scholar] [CrossRef]
- Tongo, M.; Dorfman, J.R.; Abrahams, M.R.; Mpoudi-Ngole, E.; Burgers, W.A.; Martin, D.P. Near full-length HIV type 1M genomic sequences from Cameroon: Evidence of early diverging under-sampled lineages in the country. Evol. Med. Public Health 2015, 2015, 254–265. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, M.A.; Wilkinson, E.; Vallari, A.; McArthur, C.; Sthreshley, L.; Brennan, C.A.; Cloherty, G.; de Oliveira, T. Sensitive next-generation sequencing method reveals deep genetic diversity of HIV-1 in the Democratic Republic of the Congo. J. Virol. 2017, 91, e01841-16. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Gaschen, B.; Yusim, K.; Thakallapally, R.; Kesmir, C.; Detours, V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 2001, 58, 19–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiwanuka, N.; Laeyendecker, O.; Quinn, T.C.; Wawer, M.J.; Shepherd, J.; Robb, M.; Kigozi, G.; Kagaayi, J.; Serwadda, D.; Makumbi, F.E.; et al. HIV-1 subtypes and differences in heterosexual HIV transmission among HIV-discordant couples in Rakai, Uganda. AIDS 2009, 23, 2479–2484. [Google Scholar] [CrossRef] [PubMed]
- Venner, C.M.; Nankya, I.; Kyeyune, F.; Demers, K.; Kwok, C.; Chen, P.L.; Rwambuya, S.; Munjoma, M.; Chipato, T.; Byamugisha, J.; et al. Infecting HIV-1 subtype predicts disease progression in women of Sub-Saharan Africa. EBioMedicine 2016, 13, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebit, D.M.; Arts, E.J. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis. 2011, 11, 45–56. [Google Scholar] [CrossRef]
- Oberle, C.S.; Joos, B.; Rusert, P.; Campbell, N.K.; Beauparlant, D.; Kuster, H.; Weber, J.; Schenkel, C.D.; Scherrer, A.U.; Magnus, C.; et al. Tracing HIV-1 transmission: Envelope traits of HIV-1 transmitter and recipient pairs. Retrovirology 2016, 13, 62. [Google Scholar] [CrossRef] [Green Version]
- Bártolo, I.; Camacho, R.; Barroso, H.; Bezerra, V.; Taveira, N. Rapid clinical progression to AIDS and death in a persistently seronegative HIV-1 infected heterosexual young man. AIDS 2009, 23, 2359–2362. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No. | Accession Number | Rega 2.0 | Rega 3.0 | Comet 1.0 | jpHMM | SCUEAL | Stanford | Geno2pheno 3.3 |
|---|---|---|---|---|---|---|---|---|
| 1 | AJ971094 | NA | 02_AG | 02_AG | A1, G | 02_AG-like | 02_AG/02_AG | 02_AG |
| 2 | AJ971095 | NA | 02_AG | 02_AG | A1, G | 02_AG-like/complex | 02_AG/02_AG | 02_AG |
| 3 | AJ971100 | F (F1) | F (F1) | F1 | F1 | F1 | D/F | F1 |
| 4 | AJ971096 | NA | 02_AG | 02_AG | A1, G | 02_AG-like | 02_AG/02_AG | 02_AG |
| 5 | AJ971133 | NA | B-like | B | B | B, D recombinant/B | B/B | B |
| 6 | GQ399318 | B | Recombinant of B, D | B | B | B | B/B | B |
| 7 | AM113750 | NA | 02_AG | 02_AG | A1, G | complex | 02_AG/02_AG | 02_AG |
| 8 | JX046417 | A (A1) | A (A1) | A1 | A1 | A-ancestral, A1 rec/A, A1 recombinant/A1 | A/CRF01_AE | 01_AE |
| 9 | JX046416 | A (A1) | A (01_AE) | 01_AE | 01_AE | AE | 01_AE/01_AE | 01_AE |
| 10 | JX046415 | NA | 02_AG | 02_AG | A1, G | complex | 02_AG/02_AG | 02_AG |
| 11 | JX046413 | A (A1) | A (A1) | A1 | A1 | complex/A, A-ancestral recombinant | A/01_AE | 01_AE |
| 12 | JX028352 | B | NA (recombinant of B, D) | B | B | B | B/B | B |
| 13 | JX046414 | 19_cpx | 19_cpx | unassigned; D, 11_cpx, G, 20_BG | D | complex | D/D | D |
| 14 | JX028338 | NA | B | B | B | B | B/B | B |
| 15 | JX046406 | D | D | D | B, D | D | D/D | D |
| 16 | JX046405 | D | D | D | B, D | D | D/D | D |
| 17 | JX028323 | NA | B | B | B | B | B/B | B |
| 18 | JX046407 | NA | B, potential recombinant | B | B | B | B/B | B |
| 19 | JX046408 | NA | Recombinant of F1, 05_DF | unassigned; B, F1 | B, F1 | B, F1 recombinant | B/F | F1 |
| 20 | JX046409 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 21 | JX046410 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 22 | KP013668 | G | G | unassigned; 18_cpx, 02_AG, G | G | G | G/G | 02_AG |
| 23 | JX046412 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 24 | JX046404 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 25 | JX046411 | A (A1) | A (A1) | A1 | A1 | A1 | 01_AE/01_AE | 01_AE |
| 26 | KP013669 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 27 | JX028308 | B | NA, Recombinant of B, D | B | B | B | B/B | B |
| 28 | JX046403 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 29 | KF753740 | NA | Recombinant of B, D | B | B | B | B/B | B |
| 30 | KF753737 | NA | Recombinant of B, D | B | B | B | B/B | B |
| 31 | KF753747 | NA | C, potential recombinant | unassigned; F1, A1, 01_AE, B, C, 60_BC | A1, B, C | complex | C/D | C |
| 32 | KF753751 | A (A1) | A (A1) | A1 | A1 | A1 | 01_AE/01_AE | 01_AE |
| 33 | KF753741 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 34 | KF753731 | G | G | unassigned; D, J, G | G, J | AE, G recombinant | A/G | G |
| 35 | KF753711 | B | B | unassigned; B-D | B | B | B/B | B |
| 36 | KP013665 | B | B | B | B | B, D recombinant/B | B/B | B |
| 37 | KF753704 | NA | NA (G, B) | G (check for 02_AG) | A1, B, G | complex | 02_AG/B | 02_AG |
| 38 | KF753709 | NA | B | B | B | B | B/B | B |
| 39 | KF753699 | A (A1) | A (01_AE) | 01_AE | 01_AE | 15_01B/A1, AE recombinant/AE, F-ancestral recombinant | 01_AE/01_AE | 01_AE |
| 40 | KP013656 | B | B | B (check for 29_BF) | B | B | B/B | B |
| 41 | KP013648 | A (A1) | A (A1) | A1 | A1 | A1 | 01_AE/A | A1 |
| 42 | KP013643 | NA | B-like | B | B | B | B/B | B |
| 43 | KP013657 | NA | Recombinant of B, D | B | B | B | B/B | B |
| 44 | KP013641 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 45 | KP013660 | A (A1) | A (A1) | unassigned; D, 10_CD, 01_AE, B | 01_AE | A1, AE recombinant | 01_AE/01_AE | 01_AE |
| 46 | KP013644 | NA | B | B | B | B | B/B | B |
| 47 | KY656620 | B | B-like | B | B | B | B | B |
| 48 | KY656621 | NA | B-like | B | B | B | B | B |
| 49 | MN736711 | B | B | B | B | B, C recombinant | B | B |
| 50 | KY656626 | A (A1) | A (A1) | A1 | A1 | A1 | A (A_FSU) | 01_AE |
| 51 | KY656630 | NA | 02_AG | 02_AG | A1, G | A4, G recombinant | 02_AG | 02_AG |
| 52 | KY656636 | A (A1) | A (A1) | A1 | A1 | A1 | A (A_FSU) | 01_AE |
| 53 | KY656616 | NA | B-like | B | B | B | B | B |
| 54 | KY656639 | NA | G (02_AG) | 02_AG | F1, G | G | 02_AG | 02_AG |
| 55 | KY656643 | A (A1) | A (A1) | A1 | A1 | A1 | A | 01_AE |
| 56 | KY656648 | NA | Recombinant of B, D | B | B | B | B | B |
| 57 | KY656649 | B | B | B | B | B, F1 recombinant | B | B |
| 58 | KY656650 | B | Recombinant of B, D | B | B | B | B | B |
| 59 | KY656640 | A (01_AE) | A (01_AE) | 01_AE (check for 15_01B) | 01_AE | AE | 01_AE | 01_AE |
| 60 | KY656656 | NA | B-like | B | B | B | B | B |
| 61 | KY656657 | A (A1) | A (A1) | A1 | A1 | A1 | A, 01_AE | 01_AE |
| 62 | KY656658 | A (A1) | A (A1) | A1 | A1 | A1 | A, 01_AE | 01_AE |
| 63 | KY656659 | A (A1) | A (A1) | A1 | A1 | A1 | A, 01_AE | 01_AE |
| 64 | MF987697 | NA | Recombinant of B, D | B | B | B | B | B |
| 65 | KY656670 | NA | Recombinant of B, D | B | B | B | B | B |
| 66 | KY656671 | A (A1) | A (A1) | A1 | A1 | A1 | A/01_AE | 01_AE |
| 67 | MF987719 | NA | NA | unassigned, B–G | D | complex | D | D |
| 68 | MF987720 | NA | NA | unassigned, B–G | D | complex | D | D |
| No. | Accession Number | Genome Sequence Range 1 | Rega 3.0 | Comet | jpHMM | SimPlot | Final Result |
|---|---|---|---|---|---|---|---|
| 11 | MN736709 | 497–7842 | A (A1) | A1 | A1, K, A1 | A1/K or 45_cpx | A|A/U|A|K|A |
| 12 | MN736698 | 497–9496 | B | B | B | B | B |
| 25 | MN736699 | 497–9487 | A (A1) | A1 | A1 | A6 | A6 |
| 29 | MN736700 | 497–9496 | B | B | B | B | B |
| 31 | MN736710 | 497–7843 | C | unassigned; 60_BC, B, 82_cpx, 01_AE, 58_01B, 01_AE, 58_01B, 01_AE, 58_01B, 59_01B, 58_01B, 59_01B, 58_01B, 59_01B, 01_AE, 59_01B, 58_01B, 02_AG, 58_01B, 02_AG, A1, 69_01B, 59_01B, 69_01B, 53_01B, 69_01B, 53_01B, 69_01B, 55_01[..] | C, B, C, 01_AE, C | 60_BC | 60_BC|01_AE|60_BC |
| 34 | MN736701 | 497–9496 | recombinant of 13_cpx, G, A1, J, H | unassigned; G, A1, 01_AE, D, J, C, A2, C, B, D, B, D, B, D, J, C, A1, 01_AE | A1, G, D, J, B, J, G, A1, 01_AE | complex | U|A|G|U|G|J| G|U|J|U|G| A|01_AE |
| 37 | MN736702 | 497–9433 | recombinant of 02_AG, B, G, A1 | unassigned; 02_AG, 20_BG, 56_cpx, 20_BG, 56_cpx, 90_BF1, B, 56_cpx, B, 56_cpx, B, 71_BF1, 51_01B, 71_BF1, 39_BF, B, 69_01B, B, 69_01B, B, 69_01B, B, 56_cpx, 19_cpx, 24_BG, 19_cpx, 56_cpx, 24_BG, 56_cpx, G, 14_BG, 43_02G, G, 43_02G[..] | A1, B, A1, G, B, A1, G, B, A1, B, G | 56_cpx, B | 56_cpx|B|56_cpx |
| 39 | MN736703 | 497–9496 | 01_AE | unassigned; 01_AE, B, D | 01_AE | 01_AE | 01_AE |
| 43 | MN736704 | 497–9412 | B | unassigned; B, C, G | B | B, B/D, B, B/D | B |
| 54 | MN736705 | 497–9445 | 02_AG | 02_AG | A1, G, A1, G, A1, G, A1, G, A1 | 02_AG | 02_AG|A|02_AG |
| 56 | MN736706 | 497–9496 | B | B | B | B | B |
| 65 | MN736707 | 497–9496 | B | B | B | B | B |
| 68 | MN736708 | 497–9496 | recombinant of G, A1, D, B, F1 | unassigned; G, D, B, D, B, D, A1 | A1, G, D, B, A1, G, A2, G, A1, G, A1, G | complex | U|19_cpx|G|19_cpx| B|19_cpx|A|19_cpx|U|19_cpx|G|19_cpx|G|19_cpx|G |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunar, M.M.; Mlakar, J.; Zorec, T.M.; Poljak, M. HIV-1 Unique Recombinant Forms Identified in Slovenia and Their Characterization by Near Full-Length Genome Sequencing. Viruses 2020, 12, 63. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010063
Lunar MM, Mlakar J, Zorec TM, Poljak M. HIV-1 Unique Recombinant Forms Identified in Slovenia and Their Characterization by Near Full-Length Genome Sequencing. Viruses. 2020; 12(1):63. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010063
Chicago/Turabian StyleLunar, Maja M., Jana Mlakar, Tomaž Mark Zorec, and Mario Poljak. 2020. "HIV-1 Unique Recombinant Forms Identified in Slovenia and Their Characterization by Near Full-Length Genome Sequencing" Viruses 12, no. 1: 63. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010063