JCPyV T-Antigen Activation of the Anti-Apoptotic Survivin Promoter—Its Role in the Development of Progressive Multifocal Leukoencephalopathy

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
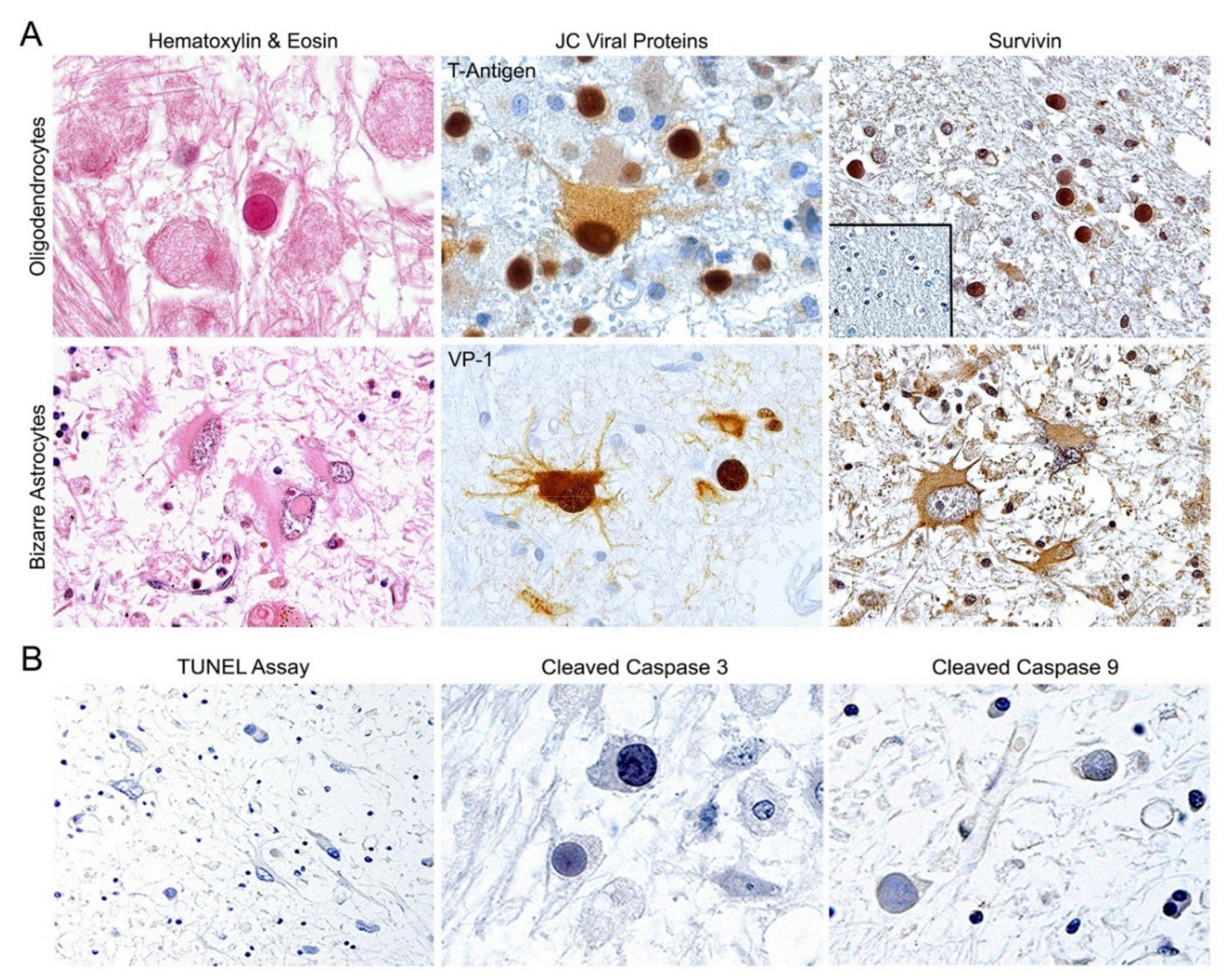
3.1. Expression of Survivin But Not Apoptotic Proteins in JCPyV-Infected Cells of PML
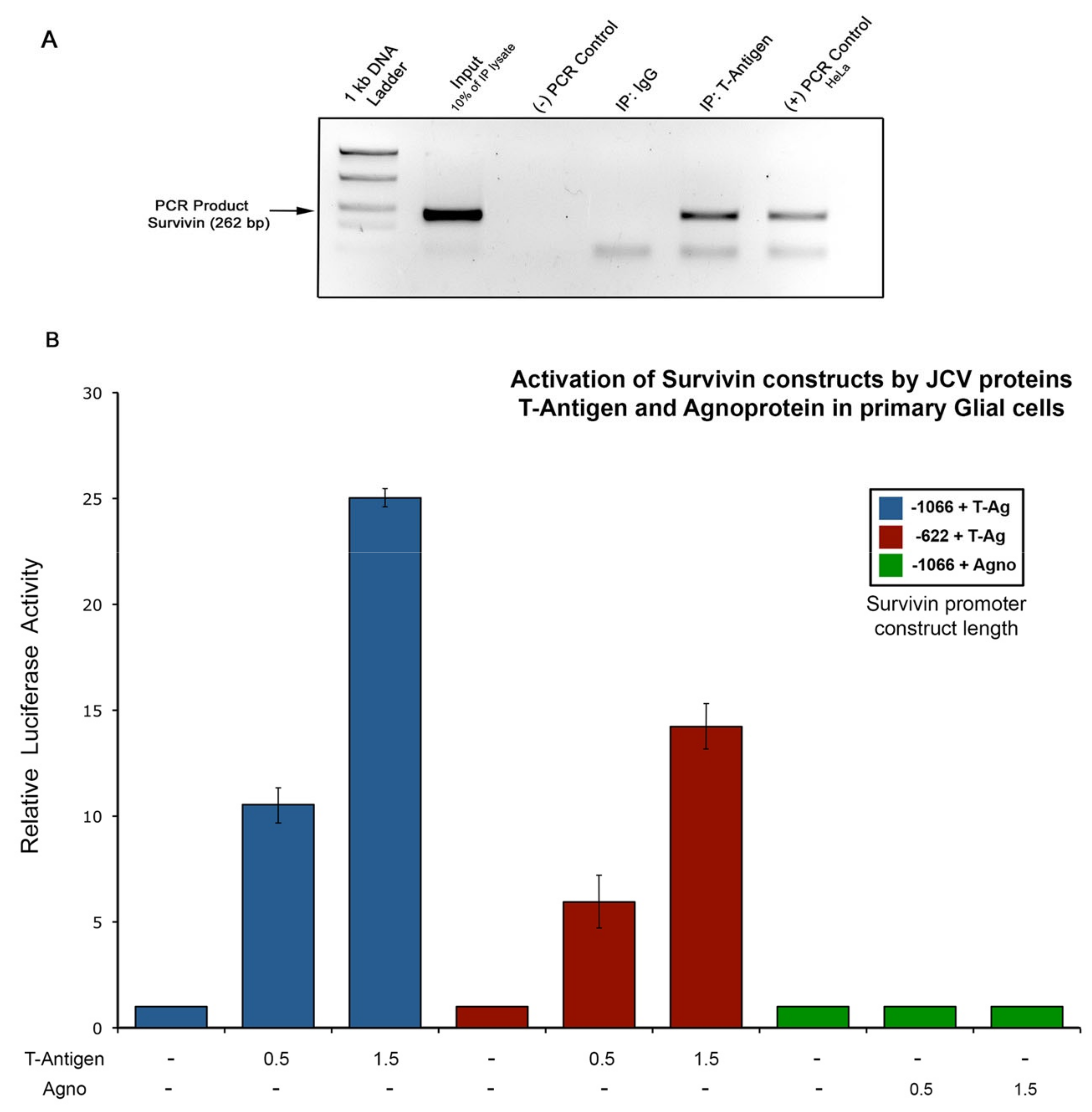
3.2. Activation of the Survivin Promoter by JCPyV T-Antigen
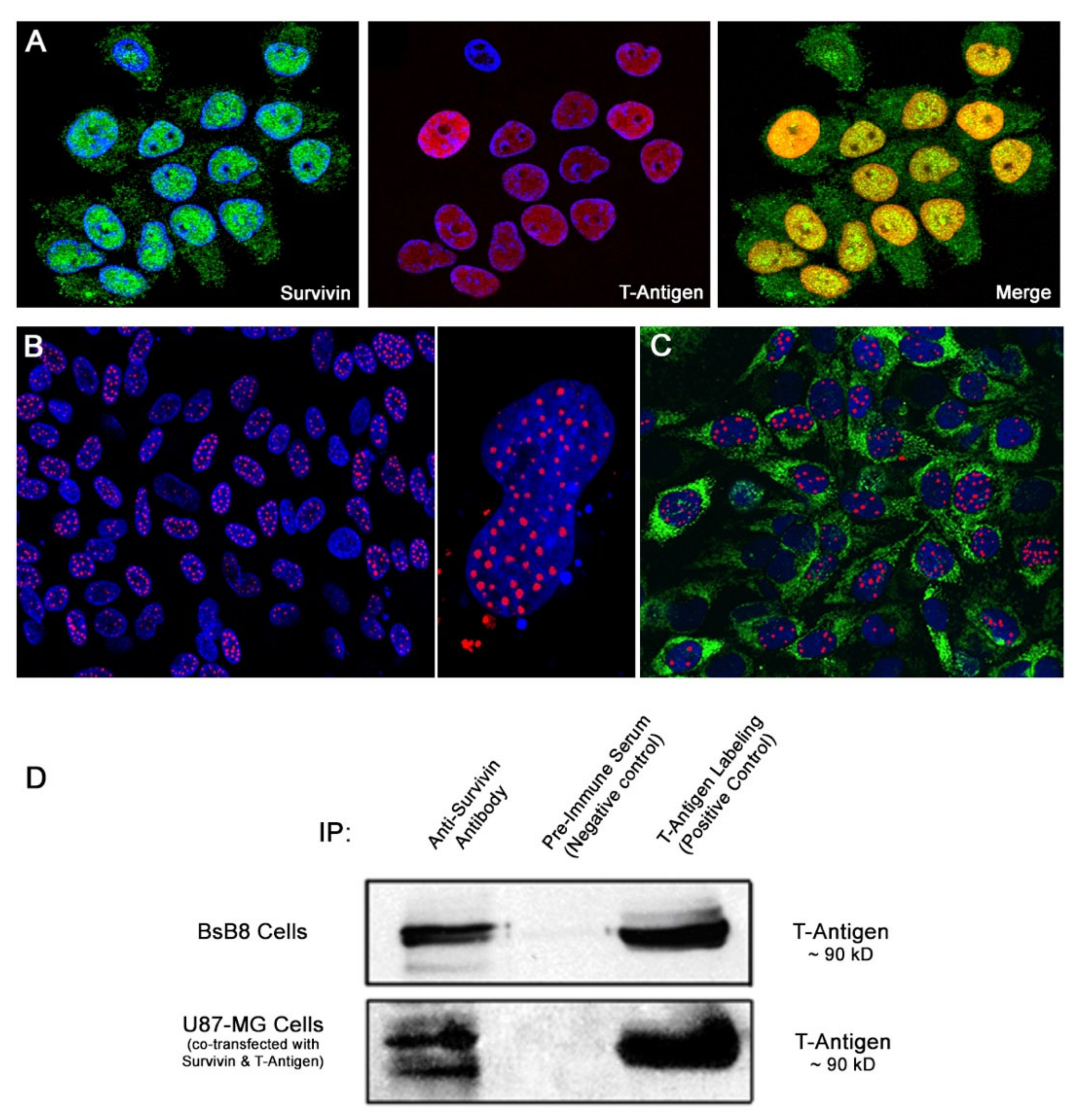
3.3. Co-Localization and Physical Interaction between T-Antigen and Survivin in Cell Cultures
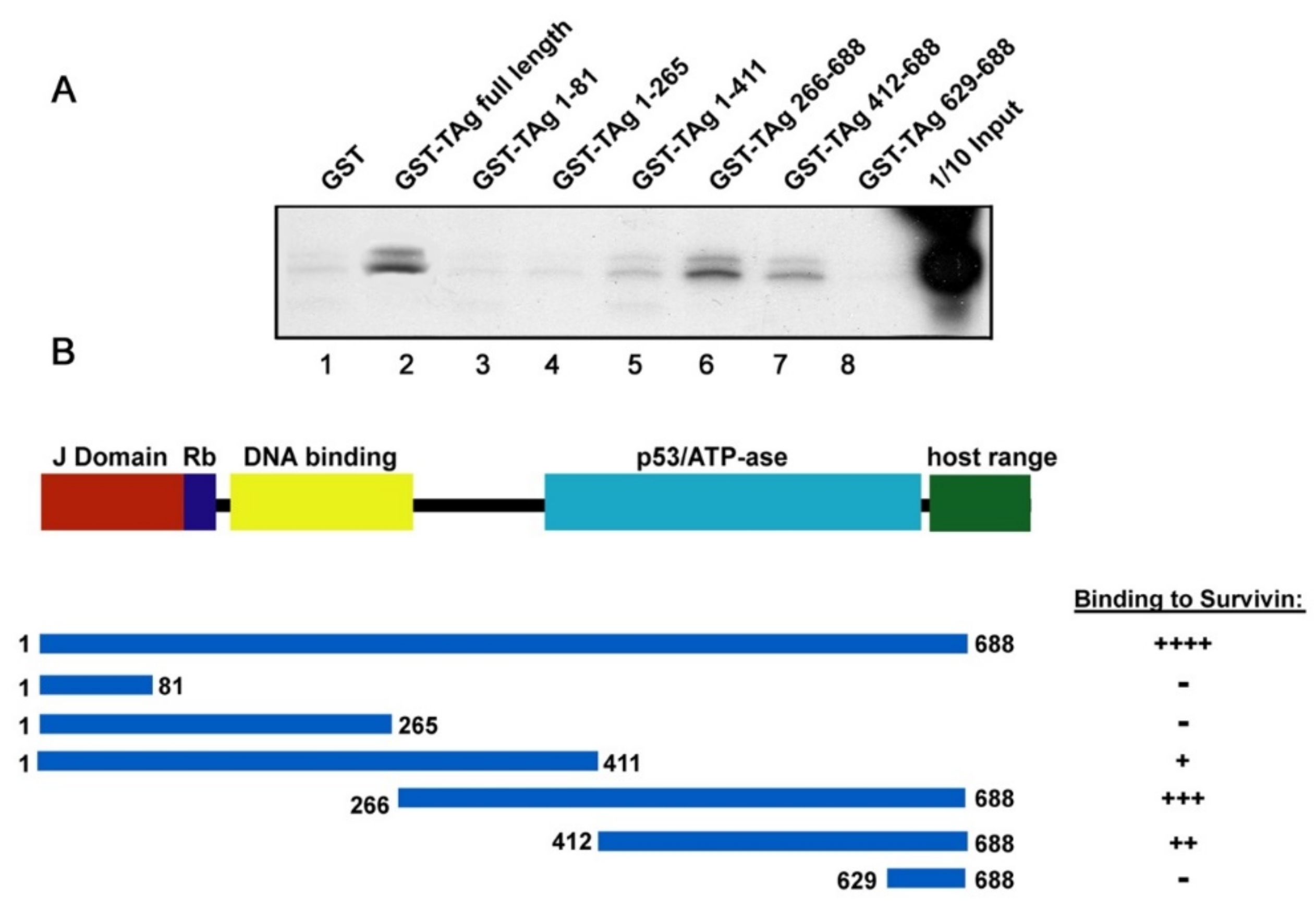
3.4. Binding of JCPyV T-Antigen to Survivin
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B. ICTV Virus taxonomy profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R.; Concha, M. Progressive Multifocal Leukoencephalopathy: The evolution of a disease once considered rare. J. Neurovirol. 1995, 1, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Piña-Oviedo, S. HIV disorders of the brain: Pathology and pathogenesis. Front. Biosci. 2006, 11, 718–732. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Ho, D.D.; Abdool Karim, Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet 2006, 368, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.R. Progressive Multifocal Leukoencephalopathy and newer biological agents. Drug Saf. Int. J. Med. Toxicol. Drug Exp. 2010, 33, 969–983. [Google Scholar] [CrossRef]
- Major, E.O. Progressive Multifocal Leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med. 2010, 61, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Åström, K.E.; Mancall, E.L.; Richardson, E.P., Jr. Progressive Multifocal Leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin’s disease. Brain 1958, 81, 93–111. [Google Scholar] [CrossRef]
- Del Valle, L.; Pina-Oviedo, S. Human Polyomavirus JCPyV and Its role in Progressive Multifocal Leukoencephalopathy and oncogenesis. Front. Oncol. 2019, 9, 711. [Google Scholar] [CrossRef] [Green Version]
- Shankar, S.K.; Mahadevan, A.; Kovoor, J.M. Neuropathology of viral infections of the Central Nervous System. Neuroimaging Clin. N. Am. 2008, 18, 19–39. [Google Scholar] [CrossRef]
- Aurelian, L. HSV-induced apoptosis in herpes encephalitis. Curr. Top. Microbiol. Immunol. 2005, 289, 79–111. [Google Scholar] [CrossRef]
- DeBiasi, R.L.; Kleinschmidt-DeMasters, B.K.; Richardson-Burns, S.; Tyler, K.L. Central Nervous System apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J. Infect. Dis. 2002, 186, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- McQuaid, S.; McMahon, J.; Herron, B.; Cosby, S.L. Apoptosis in measles virus-infected human Central Nervous System tissues. Neuropathol. Appl. Neurobiol. 1997, 23, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Arai, N.; Satoh, J.; Suzuki, H.; Katayama, K.; Tamagawa, K.; Morimatsu, Y. Neurodegenerative mechanisms in subacute sclerosing panencephalitis. J. Child Neurol. 2002, 17, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Blondel, B.; Colbere-Garapin, F.; Couderc, T.; Wirotius, A.; Guivel-Benhassine, F. Poliovirus, pathogenesis of poliomyelitis, and apoptosis. Curr. Top. Microbiol. Immunol. 2005, 289, 25–56. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, L.; Tseggai, T.; Dhingra, V.; Fu, Z.F. Rabies virus-induced apoptosis involves caspase-dependent and caspase-independent pathways. Virus Res. 2006, 121, 144–151. [Google Scholar] [CrossRef]
- Garden, G.A.; Budd, S.L.; Tsai, E.; Hanson, L.; Kaul, M.; D’Emilia, D.M.; Friedlander, R.M.; Yuan, J.; Masliah, E.; Lipton, S.A. Caspase cascades in Human Immunodeficiency Virus-associated neurodegeneration. J. Neurosci. 2002, 22, 4015–4024. [Google Scholar] [CrossRef] [Green Version]
- Gray, F.; Adle-Biassette, H.; Brion, F.; Ereau, T.; le Maner, I.; Levy, V.; Corcket, G. Neuronal apoptosis in human immunodeficiency virus infection. J. Neurovirol. 2000, 6 (Suppl. 1), S38–S43. [Google Scholar]
- Kolson, D.L.; Sabnekar, P.; Baybis, M.; Crino, P.B. Gene expression in TUNEL-positive neurons in human immunodeficiency virus-infected brain. J. Neurovirol. 2004, 10 (Suppl. 1), 102–107. [Google Scholar] [CrossRef]
- Joo, C.H.; Kim, Y.K.; Lee, H.; Hong, H.; Yoon, S.Y.; Kim, D. Coxsackievirus B4-induced neuronal apoptosis in rat cortical cultures. Neurosci. Lett. 2002, 326, 175–178. [Google Scholar] [CrossRef]
- Feuer, R.; Mena, I.; Pagarigan, R.R.; Harkins, S.; Hassett, D.E.; Whitton, J.L. Coxsackievirus B3 and the neonatal CNS: The roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am. J. Pathol. 2003, 163, 1379–1393. [Google Scholar] [CrossRef]
- Swarup, V.; Ghosh, J.; Duseja, R.; Ghosh, S.; Basu, A. Japanese encephalitis virus infection decrease endogenous IL-10 production: Correlation with microglial activation and neuronal death. Neurosci. Lett. 2007, 420, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Boldorini, R.; Cristina, S.; Vago, L.; Tosoni, A.; Guzzetti, S.; Costanzi, G. Ultrastructural studies in the lytic phase of Progressive Multifocal Leukoencephalopathy in AIDS patients. Ultrastruct. Pathol. 1993, 17, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Seth, P.; Diaz, F.; Tao-Cheng, J.H.; Major, E.O. JC Virus induces non-apoptotic cell death of human central nervous system progenitor cell-derived astrocytes. J. Virol. 2004, 78, 4884–4891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piña-Oviedo, S.; Urbanska, K.; Radhakrishnan, S.; Sweet, T.; Reiss, K.; Khalili, K.; Del Valle, L. Effects of JC Virus infection on anti-apoptotic protein Survivin in Progressive Multifocal Leukoencephalopathy. Am. J. Pathol. 2007, 170, 1291–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Brattain, M.G. Role of the Survivin gene in pathophysiology. Am. J. Pathol. 2006, 169, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosini, G.; Adida, C.; Sirugo, G.; Altieri, D.C. Induction of apoptosis and inhibition of cell proliferation by Survivin gene targeting. J. Biol. Chem. 1998, 273, 11177–11182. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by Survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef]
- Altieri, D.C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Altieri, D.C. Transcriptional analysis of human Survivin gene expression. Biochem. J. 1999, 344, 305–311. [Google Scholar] [CrossRef]
- Wagner, M.; Schmelz, K.; Dorken, B.; Tamm, I. Transcriptional regulation of human Survivin by early growth response (Egr)-1 transcription factor. Int. J. Cancer 2008, 122, 1278–1287. [Google Scholar] [CrossRef]
- Torres, V.A.; Tapia, J.C.; Rodriguez, D.A.; Lladser, A.; Arredondo, C.; Leyton, L.; Quest, A.F. E-cadherin is required for caveolin-1-mediated down-regulation of the inhibitor of apoptosis protein Survivin via reduced beta-catenin-Tcf/Lef-dependent transcription. Mol. Cell. Biol. 2007, 27, 7703–7717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqa, A.; Long, L.M.; Li, L.; Marciniak, R.A.; Kazhdan, I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer 2008, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Chiang, K.Y.; Zhu, N.; Findley, H.W.; Zhou, M. Contribution of STAT3 to the activation of Survivin by GM-CSF in CD34+ cell lines. Exp. Hematol. 2007, 35, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Apontes, P.; Song, L.; Liang, P.; Yang, L.; Li, F. Molecular mechanism of upregulation of Survivin transcription by the AT-rich DNA-binding ligand, Hoechst33342: Evidence for Survivin involvement in drug resistance. Nucleic Acids Res. 2007, 35, 2390–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.H.; Karna, P.; Cao, Z.; Jiang, B.H.; Zhou, M.; Yang, L. Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1alpha signal pathways increases resistance to apoptosis by up-regulating Survivin gene expression. J. Biol. Chem. 2006, 281, 25903–25914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xie, M.; Yang, J.; Yang, D.; Deng, R.; Wan, Y.; Yan, B. The expression of antiapoptotic protein Survivin is transcriptionally upregulated by DEC1 primarily through multiple sp1 binding sites in the proximal promoter. Oncogene 2006, 25, 3296–3306. [Google Scholar] [CrossRef] [Green Version]
- Bao, R.; Connolly, D.C.; Murphy, M.; Green, J.; Weinstein, J.K.; Pisarcik, D.A.; Hamilton, T.C. Activation of cancer-specific gene expression by the Survivin promoter. J. Natl. Cancer Inst. 2002, 94, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Yang, J.; Ramnath, N.; Javle, M.M.; Tan, D. Nuclear or cytoplasmic expression of Survivin: What is the significance? Int. J. Cancer 2005, 114, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Altieri, D.C. Validating Survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Hino, R.; Uozaki, H.; Inoue, Y.; Shintani, Y.; Ushiku, T.; Sakatani, T.; Takada, K.; Fukayama, M. Survival advantage of EBV-associated gastric carcinoma: Survivin up-regulation by viral latent membrane protein 2A. Cancer Res. 2008, 68, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Borbely, A.A.; Murvai, M.; Konya, J.; Beck, Z.; Gergely, L.; Li, F.; Veress, G. Effects of human papillomavirus type 16 oncoproteins on Survivin gene expression. J. Gen. Virol. 2006, 87, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, N.; Yin, L.; Cai, N.; Ma, H.; You, J.; Zhang, H.; Wang, H.; He, R.; Ye, L. Hepatitis B virus X protein upregulates Survivin expression in hepatoma tissues. J. Med. Virol. 2005, 77, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Marusawa, H.; Matsuzawa, S.; Welsh, K.; Zou, H.; Armstrong, R.; Tamm, I.; Reed, J.C. HBXIP functions as a cofactor of Survivin in apoptosis suppression. EMBO J. 2003, 22, 2729–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Roshal, M.; Li, F.; Blackett, J.; Planelles, V. Upregulation of Survivin by HIV-1 Vpr. Apoptosis 2003, 8, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Tomita, M.; Matsuda, T.; Ohta, T.; Tanaka, Y.; Fujii, M.; Hatano, M.; Tokuhisa, T.; Mori, N. Transcriptional activation of Survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int. J. Cancer 2005, 115, 967–974. [Google Scholar] [CrossRef]
- Rice, P.W.; Cole, C.N. Efficient transcriptional activation of many simple modular promoters by Simian Virus 40 large T-Antigen. J. Virol. 1993, 67, 6689–6697. [Google Scholar] [CrossRef] [Green Version]
- Taylor, I.C.; Solomon, W.; Weiner, B.M.; Paucha, E.; Bradley, M.; Kingston, R.E. Stimulation of the human heat shock protein 70 promoter in vitro by Simian Virus 40 large T-Antigen. J. Biol. Chem. 1989, 264, 16-160–16-164. [Google Scholar]
- Graham, F.L.; van der Eb, A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973, 52, 456–467. [Google Scholar] [CrossRef]
- Krynska, B.; Otte, J.; Franks, R.; Khalili, K.; Croul, S. Human ubiquitous JCV(CY) T-Antigen gene induces brain tumors in experimental animals. Oncogene 1999, 18, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Connell, C.M.; Wheatley, S.P.; McNeish, I.A. Nuclear Survivin abrogates multiple cell cycle checkpoints and enhances viral oncolysis. Cancer Res. 2008, 68, 7923–7931. [Google Scholar] [CrossRef] [Green Version]
- Connell, C.M.; Colnaghi, R.; Wheatley, S.P. Nuclear Survivin has reduced stability and is not cytoprotective. J. Biol. Chem. 2008, 283, 3289–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauber, R.H.; Mann, W.; Knauer, S.K. Nuclear and cytoplasmic Survivin: Molecular mechanism, prognostic, and therapeutic potential. Cancer Res. 2007, 67, 5999–6002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, S.; Otte, J.; Enam, S.; Del Valle, L.; Khalili, K.; Gordon, J. JC Virus-induced changes in cellular gene expression in primary human astrocytes. J. Virol. 2003, 77, 10638–10644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic Survivin gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar] [CrossRef] [Green Version]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human Survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef] [Green Version]
- Krynska, B.; Gordon, J.; Otte, J.; Franks, R.; Knobler, R.; DeLuca, A.; Giordano, A.; Khalili, K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J. Cell. Biochem. 1997, 67, 223–230. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Noch, E.K.; Khalili, K. A novel role of Rac1 GTPase in JCV T-Antigen-mediated beta-catenin stabilization. Oncogene 2007, 26, 7628–7636. [Google Scholar] [CrossRef]
- White, M.K.; Gordon, J.; Reiss, K.; Del Valle, L.; Croul, S.; Giordano, A.; Darbinyan, A.; Khalili, K. Human polyomaviruses and brain tumors. Brain Res. Brain Res. Rev. 2005, 50, 69–85. [Google Scholar] [CrossRef]
- Del Valle, L.; White, M.K.; Khalili, K. Potential mechanisms of the human Polyomavirus JC in neural oncogenesis. J. Neuropathol. Exp. Neurol. 2008, 67, 729–740. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Age | Gender | Diagnosis | Underlying Disease | Immunohistochemistry | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| VP-1 | T-Antigen | Survivin | C/Caspase-3 | C/Caspase 9 | TUNEL Assay | |||||
| 1 | 44 y/o | Male | AIDS-PML | HIV+/AIDS | ++++ | +++ | ++++ | - | - | - |
| 2 | 6 y/o | Male | AIDS-PML | HIV+/AIDS | +++ | +++ | ++++ | - | - | - |
| 3 | 48 y/o | Male | AIDS-PML | HIV+/AIDS | ++++ | ++++ | +++ | - | - | - |
| 4 | 36 y/o | Male | AIDS-PML | HIV+/AIDS | +++ | +++ | ++++ | - | - | - |
| 5 | 33 y/o | Male | AIDS-PML | AIDS + CNS Lymphoma | ++++ | +++ | ++++ | - | - | - |
| 6 | 73 y/o | Female | Non-AIDS PML | Renal Transplant | ++++ | ++++ | ++++ | - | - | - |
| 7 | 46 y/o | Male | Non-AIDS PML | Lymphocytic Leukemia | ++++ | +++ | ++++ | - | - | - |
| 8 | 31 y/o | Female | AIDS-PML | HIV+/AIDS | ++++ | ++++ | ++++ | - | - | - |
| 9 | 35 y/o | Male | AIDS-PML | HIV+/AIDS | ++++ | ++++ | ++++ | - | - | - |
| 10 | 37 y/o | Male | AIDS-PML | HIV+/AIDS | +++ | ++ | +++ | - | - | - |
| 11 | 48 y/o | Female | AIDS-PML | HIV+/AIDS | ++++ | ++++ | +++ | - | - | - |
| 12 | 44 y/o | Male | AIDS-PML | HIV+/AIDS | ++++ | ++++ | ++++ | - | - | - |
| 13 | 57 y/o | Male | AIDS-PML | HIV+/AIDS | +++ | +++ | ++++ | - | - | - |
| 14 | 35 y/o | Female | AIDS-PML | HIV+/AIDS | ++++ | ++++ | ++++ | - | - | - |
| 15 | 45 y/o | Female | Non-AIDS PML | Chron’s Dis/Natalizumab | ++++ | +++ | ++++ | - | - | - |
| 16 | 52 y/o | Male | AIDS-PML | HIV+/AIDS | ++++ | ++ | +++ | - | - | - |
| 1 | 41 y/o | Male | HIV-Encephalitis | HIV+/AIDS | - | - | - | +++ | ++ | ++++ |
| 2 | 45 y/o | Male | HIV-Encephalitis | HIV+/AIDS | - | - | - | +++ | ++ | +++ |
| 3 | 32 y/o | Male | Normal Brain | Drug Overdose | - | - | - | - | - | - |
| 4 | 75 y/o | Female | Normal Brain | Breast Cancer | - | - | - | - | - | - |
| 5 | 52 y/o | Female | Normal Brain | Myocardial Infarction | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Valle, L.; Sweet, T.; Parker-Struckhoff, A.; Perez-Liz, G.; Piña-Oviedo, S. JCPyV T-Antigen Activation of the Anti-Apoptotic Survivin Promoter—Its Role in the Development of Progressive Multifocal Leukoencephalopathy. Viruses 2020, 12, 1253. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111253
Del Valle L, Sweet T, Parker-Struckhoff A, Perez-Liz G, Piña-Oviedo S. JCPyV T-Antigen Activation of the Anti-Apoptotic Survivin Promoter—Its Role in the Development of Progressive Multifocal Leukoencephalopathy. Viruses. 2020; 12(11):1253. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111253
Chicago/Turabian StyleDel Valle, Luis, Thersa Sweet, Amanda Parker-Struckhoff, Georgina Perez-Liz, and Sergio Piña-Oviedo. 2020. "JCPyV T-Antigen Activation of the Anti-Apoptotic Survivin Promoter—Its Role in the Development of Progressive Multifocal Leukoencephalopathy" Viruses 12, no. 11: 1253. https://0-doi-org.brum.beds.ac.uk/10.3390/v12111253