Structural Basis for Inhibiting Porcine Epidemic Diarrhea Virus Replication with the 3C-Like Protease Inhibitor GC376

Abstract
:1. Introduction
2. Materials and Methods
2.1. Compound
2.2. Viruses and Cells
2.3. Expression and Purification of the PEDV 3CLpro
2.4. Förster Resonance Energy Transfer (FRET)-Based Assays for 50% Inhibitory Concentration (IC50) Measurement
2.5. Cell-Based Assays for Concentration for the 50% Maximal Effect (EC50) Measurement
2.6. Western Blot Analysis
2.6.1. Indirect Immunofluorescence Assay
2.6.2. Crystallization and Structure Determination
3. Results
3.1. Inhibitory Effects of GC376 on the PEDV 3CLpro
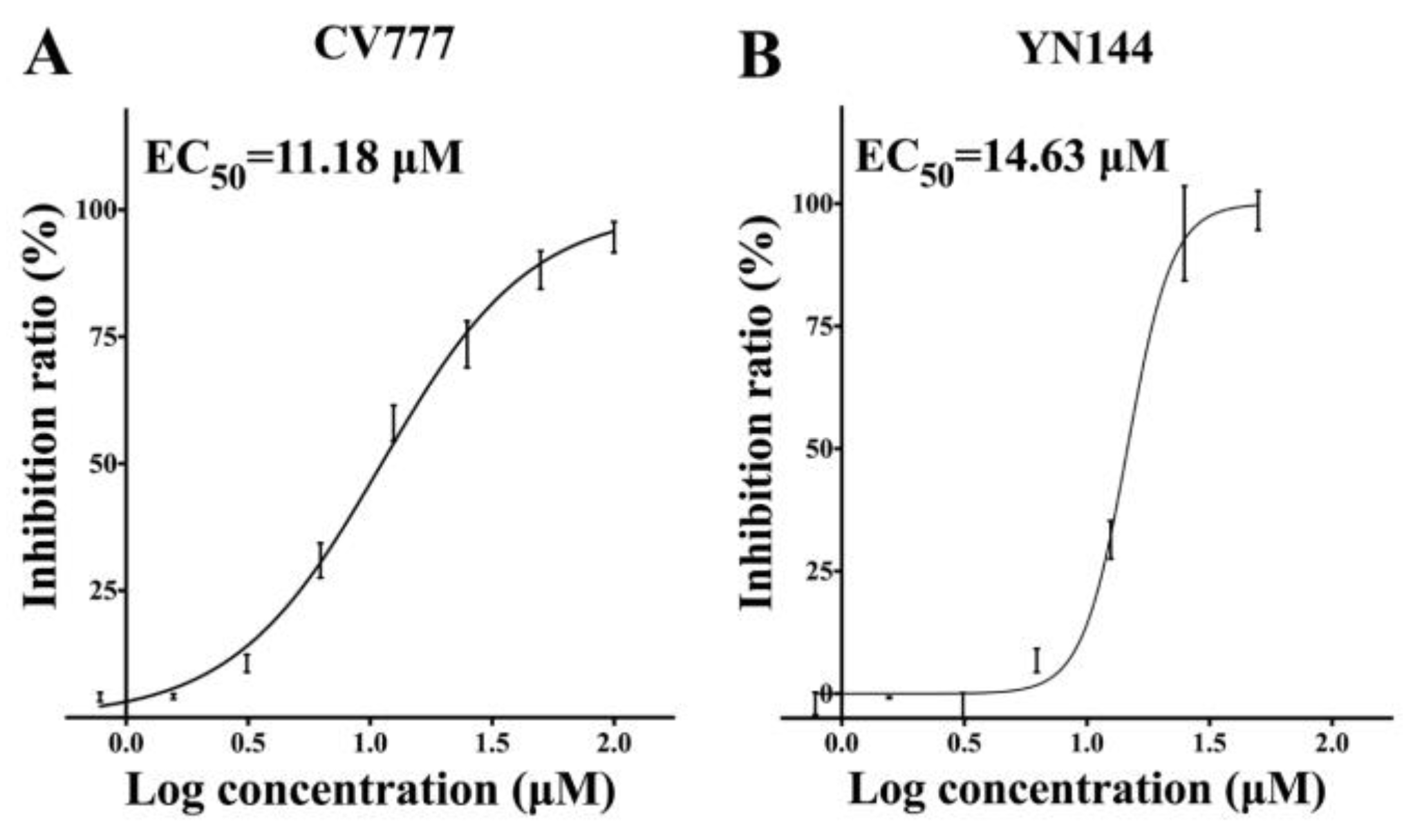
3.2. Antiviral Effects of GC376 on the PEDV CV777 and YN144 Strains
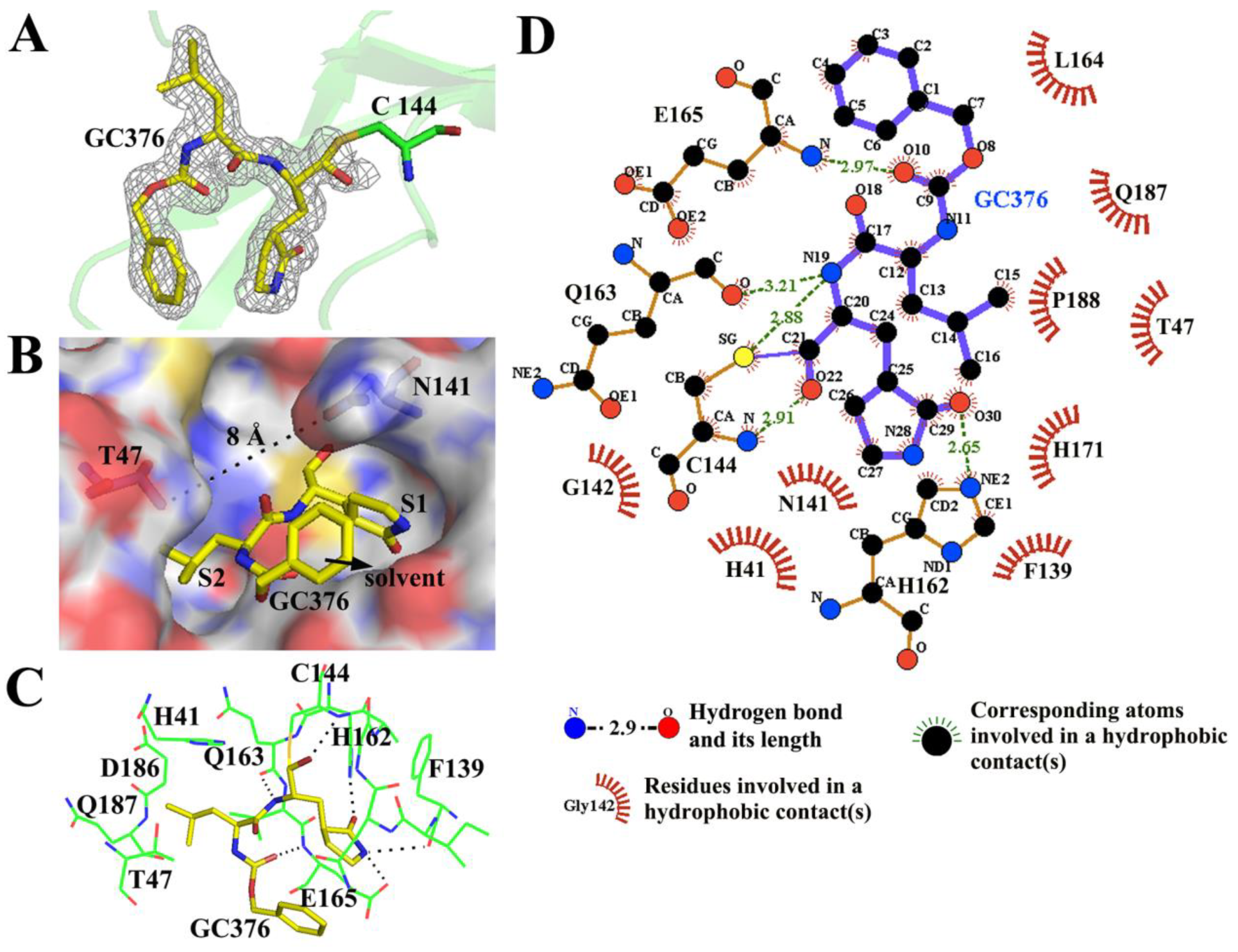
3.3. GC376 Inhibits PEDV Replication by Blocking the Catalytic Residues and Binding Pocket of 3CLpro
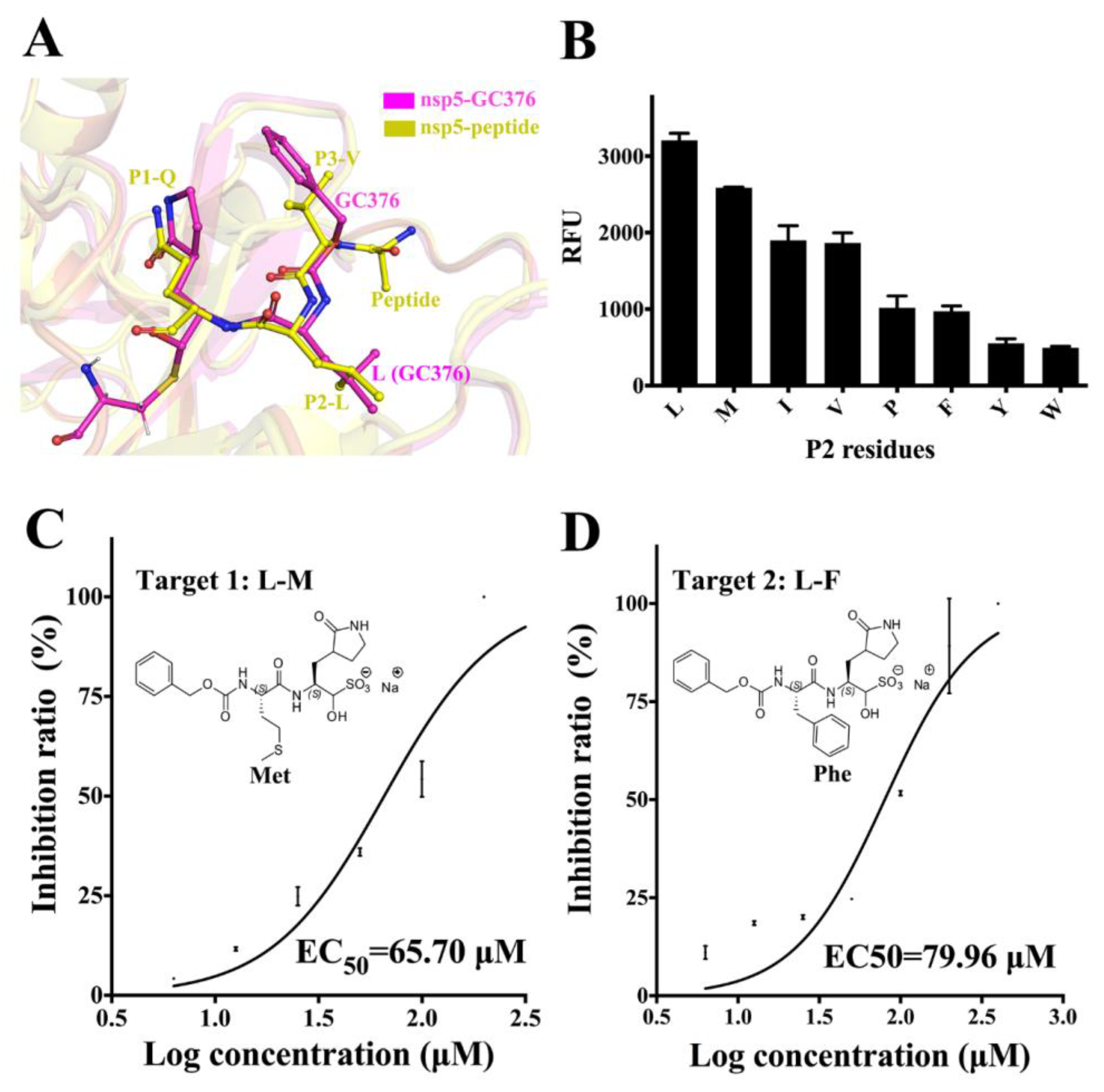
3.4. Substrate Specificity of PEDV 3CLpro at the P2 Site and the Optimization of GC376
3.5. Comparison between the PEDV 3CLpro–GC376 Complex and TGEV 3CLpro–GC376 Complex
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oldham, J. Letter to the editor. Pig Farming 1972, 10, 72–73. [Google Scholar] [CrossRef]
- Song, D.; Moon, H.; Kang, B. Porcine epidemic diarrhea: A review of current epidemiology and available vaccines. Clin. Exp. Vaccine Res. 2015, 4, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, G.W.; Hoang, H.; Schwartz, K.J.; Burrough, E.R.; Sun, D.; Madson, D.; Cooper, V.L.; Pillatzki, A.; Gauger, P.; Schmitt, B.J.; et al. Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013, 25, 649–654. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of Porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350–1353. [Google Scholar] [CrossRef]
- Sun, R.Q.; Cai, R.J.; Chen, Y.Q.; Liang, P.S.; Chen, D.K.; Song, C.X. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 2012, 18, 161–163. [Google Scholar] [CrossRef]
- Wang, X.M.; Niu, B.B.; Yan, H.; Gao, D.S.; Yang, X.; Chen, L.; Chang, H.T.; Zhao, J.; Wang, C.Q. Genetic properties of endemic Chinese Porcine epidemic diarrhea virus strains isolated since 2010. Arch. Virol. 2013, 158, 2487–2494. [Google Scholar] [CrossRef]
- Huang, Y.W.; Dickerman, A.W.; Pineyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent Porcine epidemic diarrhea virus strains in the United States. mBio 2013, 4, e00737-13. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.F.; Chen, C.C.; Moses, D.C.; Chen, Y.H.; Lin, C.H.; Tsai, Y.C.; Chou, C.Y. Porcine epidemic diarrhea virus papain-like protease 2 can be noncompetitively inhibited by 6-thioguanine. Antivir. Res. 2018, 158, 199–205. [Google Scholar] [CrossRef]
- Hegyi, A.; Friebe, A.; Gorbalenya, A.E.; Ziebuhr, J. Mutational analysis of the active centre of coronavirus 3C-like proteases. J. Gen. Virol. 2002, 83, 581–593. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Ziebuhr. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 2005, 287, 57–94. [Google Scholar]
- Thiel, V.; Herold, J.; Schelle, B.; Siddell, S.G. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 2001, 75, 6676–6681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weissbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Bacha, U.; Barrila, J.; Gabelli, S.B.; Kiso, Y.; Mario Amzel, L.; Freire, E. Development of broad-spectrum halomethyl ketone inhibitors against coronavirus main protease 3CL(pro). Chem. Biol. Drug Des. 2008, 72, 34–49. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.W.; Cheng, S.C.; Chen, W.Y.; Lin, M.H.; Chuang, S.J.; Cheng, I.H.; Sun, C.Y.; Chou, C.Y. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antivir. Res. 2015, 115, 9–16. [Google Scholar] [CrossRef]
- Kumar, V.; Shin, J.S.; Shie, J.J.; Ku, K.B.; Kim, C.; Go, Y.Y.; Huang, K.F.; Kim, M.; Liang, P.H. Identification and evaluation of potent Middle East respiratory syndrome coronavirus (MERS-CoV) 3CL(Pro) inhibitors. Antivir. Res. 2017, 141, 101–106. [Google Scholar] [CrossRef]
- Kim, Y.; Shivanna, V.; Narayanan, S.; Prior, A.M.; Weerasekara, S.; Hua, D.H.; Kankanamalage, A.C.; Groutas, W.C.; Chang, K.O. Broad-spectrum inhibitors against 3C-like proteases of feline coronaviruses and feline caliciviruses. J. Virol. 2015, 89, 4942–4950. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Liu, H.; Kankanamalage, A.C.G.; Weerasekara, S.; Hua, D.H.; Groutas, W.C.; Chang, K.O.; Pedersen, N.C. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016, 12, e1005531. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Li, C.; Li, W.; Lucio de Esesarte, E.; Guo, H.; van den Elzen, P.; Aarts, E.; van den Born, E.; Rottier, P.J.M.; Bosch, B.J. Cell attachment domains of the Porcine epidemic diarrhea virus spike protein are key targets of neutralizing antibodies. J. Virol. 2017, 91, e00273-17. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.L.; Cheng, S.C.; Shi, L.; Wang, T.Y.; Ho, K.I.; Chou, C.Y. Critical assessment of the important residues involved in the dimerization and catalysis of MERS coronavirus main protease. PLoS ONE 2015, 10, e0144865. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, 1742–1752. [Google Scholar] [CrossRef]
- Li, F. Receptor recognition and cross-species infections of SARS coronavirus. Antivir. Res. 2013, 100, 246–254. [Google Scholar] [CrossRef]
- Wu, K.; Peng, G.; Wilken, M.; Geraghty, R.J.; Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2012, 287, 8904–8911. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, C.; Du, L.; Jiang, S.; Shi, Z.; Baric, R.S.; Li, F. Two mutations were critical for bat-to-human transmission of middle east respiratory syndrome coronavirus. J. Virol. 2015, 89, 9119–9123. [Google Scholar] [CrossRef] [Green Version]
- Hulswit, R.J.; de Haan, C.A.; Bosch, B.J. Coronavirus spike protein and tropism changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Hegyi, A.; Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002, 83, 595–599. [Google Scholar] [CrossRef]
- Hsu, W.C.; Chang, H.C.; Chou, C.Y.; Tsai, P.J.; Lin, P.I.; Chang, G.G. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 2005, 280, 22741–22748. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Kuo, C.J.; Ko, T.P.; Hsu, M.F.; Tsui, Y.C.; Chang, S.C.; Yang, S.; Chen, S.J.; Chen, H.C.; Hsu, M.C.; et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 2009, 284, 7646–7655. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef] [Green Version]
- Tiew, K.C.; He, G.; Aravapalli, S.; Mandadapu, S.R.; Gunnam, M.R.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Chang, K.O.; Groutas, W.C. Design, synthesis, and evaluation of inhibitors of Norwalk virus 3C protease. Bioorg. Med. Chem. Lett. 2011, 21, 5315–5319. [Google Scholar] [CrossRef] [Green Version]
- Mandadapu, S.R.; Weerawarna, P.M.; Gunnam, M.R.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Chang, K.O.; Groutas, W.C. Potent inhibition of norovirus 3CL protease by peptidyl alpha-ketoamides and alpha-ketoheterocycles. Bioorg. Med. Chem. Lett. 2012, 22, 4820–4826. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zhu, Y.; Wu, M.; Ku, X.; Ye, S.; Li, Z.; Guo, X.; He, Q. Comparative genomic analysis of classical and variant virulent parental/attenuated strains of Porcine epidemic diarrhea virus. Viruses 2015, 7, 5525–5538. [Google Scholar] [CrossRef]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Ye, G.; Deng, F.; Shen, Z.; Luo, R.; Zhao, L.; Xiao, S.; Fu, Z.F.; Peng, G. Structural basis for the dimerization and substrate recognition specificity of Porcine epidemic diarrhea virus 3C-like protease. Virology 2016, 494, 225–235. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Kauffman, R.S.; Hurter, P.; Mueller, P. Discovery and development of telaprevir: An NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 2011, 29, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.; Venkatraman, S. The discovery and development of boceprevir: A novel, first-generation inhibitor of the hepatitis C virus NS3/4A serine protease. J. Clin. Transl. Hepatol. 2013, 1, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS (Auckland, N.Z.) 2015, 7, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Mandadapu, S.R.; Groutas, W.C.; Chang, K.O. Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antivir. Res. 2013, 97, 161–168. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PEDV 3CLpro in Complex with GC376 | |
| Data Collection | |
| Space group | P 1 21 1 |
| Cell parameter (a, b, c(Å)) | 56.86, 92.22, 58.30 |
| α, β, γ | 90.00°, 100.09°, 90.00° |
| Wavelength | 0.97918 |
| Resolution range (Å) | 46.11–1.56 |
| % Completeness | 98.6 (99.0) |
| Rmerge (last shell) | 0.055 (0.060) |
| I/σ (last shell) | 17.0 (2.5) |
| CC (1/2) | 0.999 (0.864) |
| Redundancy (last shell) | 6.9 (7.0) |
| Refinement | |
| Resolution (Å) | 39.80–1.56 |
| Rwork/Rfree | 0.174/0.207 |
| No. reflections | 90,828 |
| No of protein atoms | 8998 |
| No. of solvent atoms | 1014 |
| No. of ions/ligands | 2 |
| r.m.s.d. | |
| Bond length (Å) | 0.011 |
| Bond angle (Å) | 1.10 |
| Average B factor (Å2) | 29.34 |
| Protein | 28.75 |
| Water | 35.08 |
| Ligand | 41.97 |
| Ramachandran plot: core, allow, | 97.81%, 2.02%, |
| disallow | 0.17% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, G.; Wang, X.; Tong, X.; Shi, Y.; Fu, Z.F.; Peng, G. Structural Basis for Inhibiting Porcine Epidemic Diarrhea Virus Replication with the 3C-Like Protease Inhibitor GC376. Viruses 2020, 12, 240. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020240
Ye G, Wang X, Tong X, Shi Y, Fu ZF, Peng G. Structural Basis for Inhibiting Porcine Epidemic Diarrhea Virus Replication with the 3C-Like Protease Inhibitor GC376. Viruses. 2020; 12(2):240. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020240
Chicago/Turabian StyleYe, Gang, Xiaowei Wang, Xiaohan Tong, Yuejun Shi, Zhen F. Fu, and Guiqing Peng. 2020. "Structural Basis for Inhibiting Porcine Epidemic Diarrhea Virus Replication with the 3C-Like Protease Inhibitor GC376" Viruses 12, no. 2: 240. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020240