Antiviral Effects of Menthol on Coxsackievirus B

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture and Treatments
2.2. Generation of Coxsackievirus B3 Expressing Enhanced Green Fluorescent Protein
2.3. Western Blots
2.4. Cell Immunostaining
2.5. RNA Isolation and Quantitative PCR
2.6. Mouse Treatments
2.7. Plaque Assays
2.8. Histology
3. Results
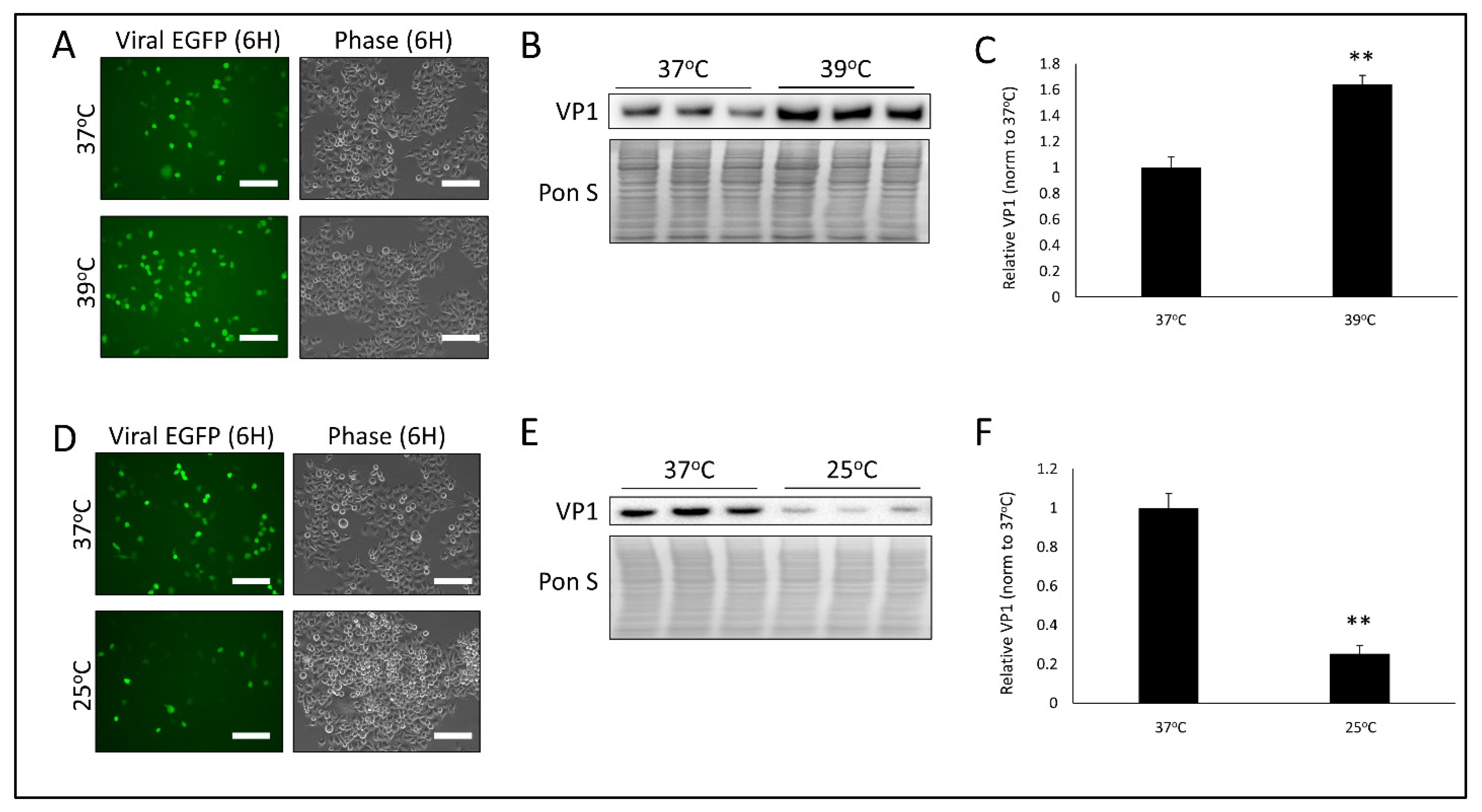
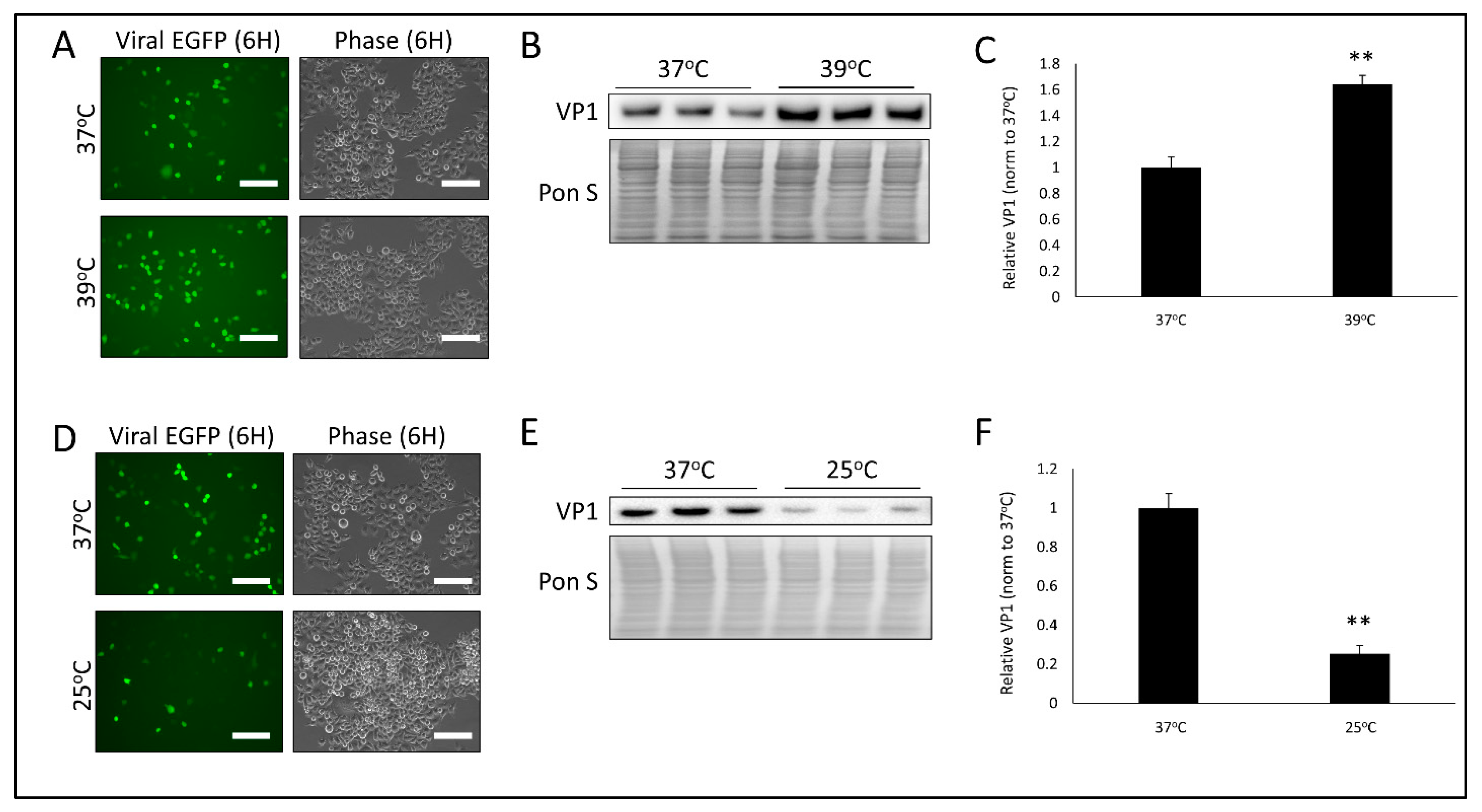
3.1. Temperature Significantly Alters CVB Infection
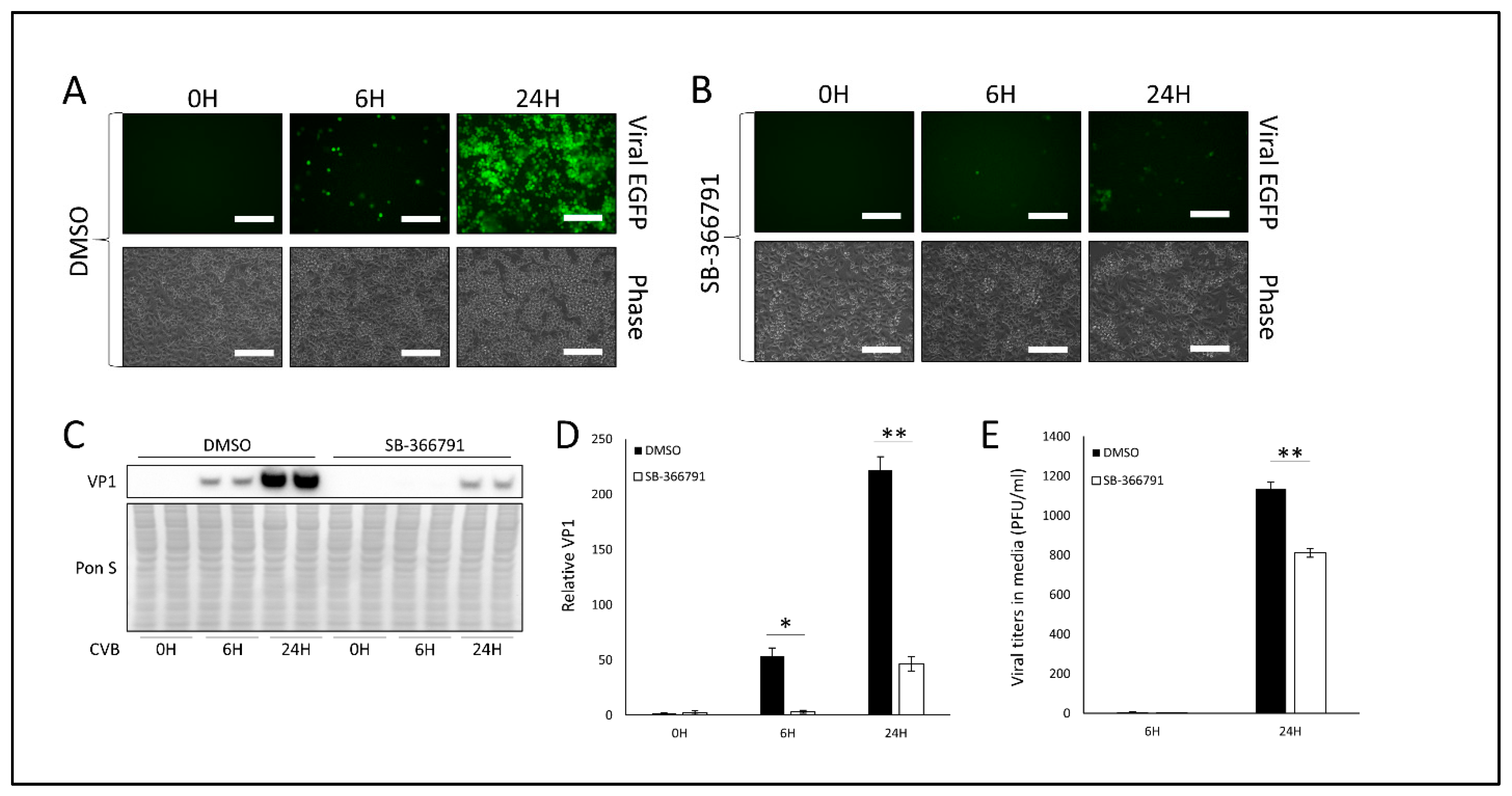
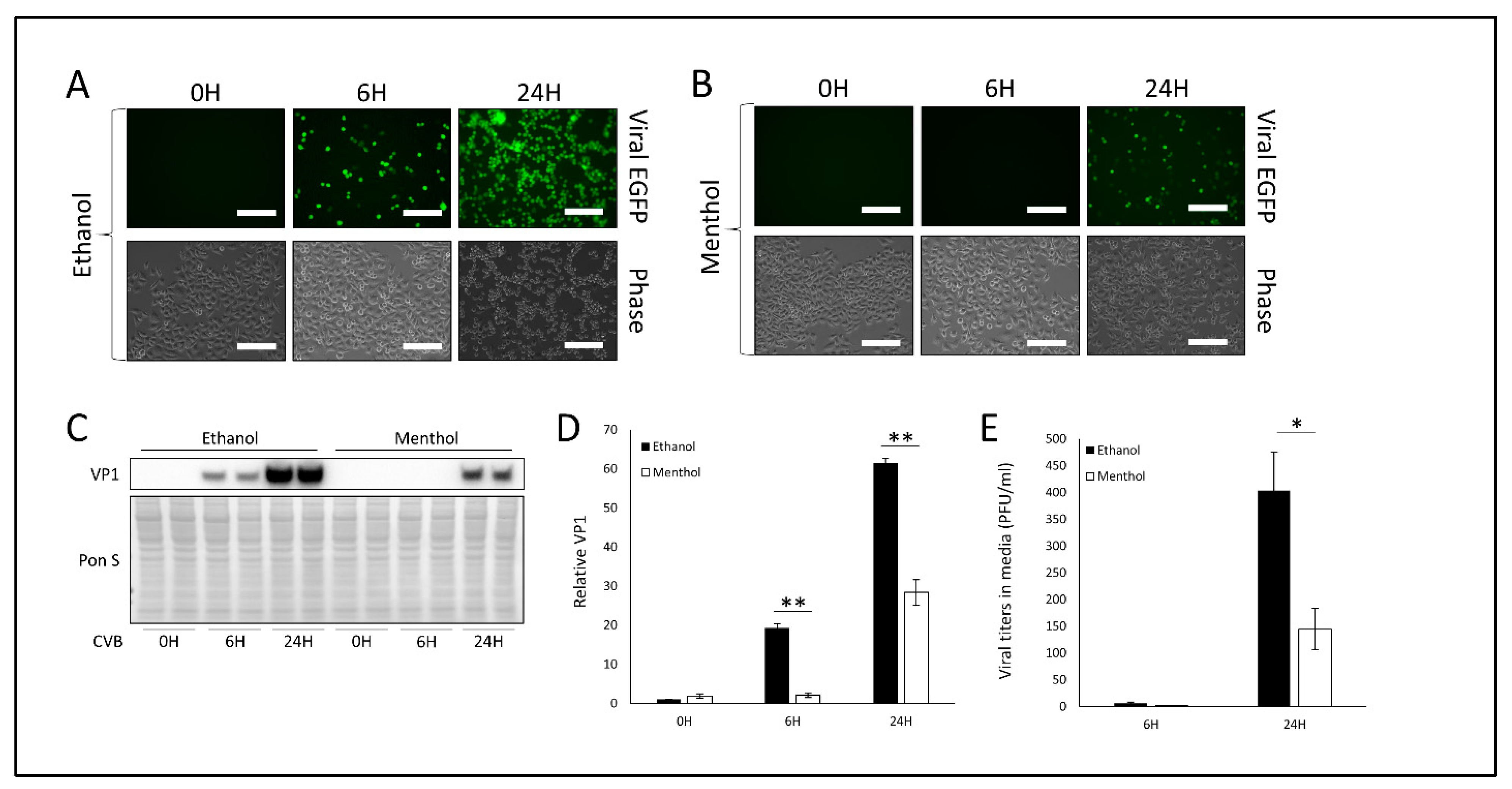
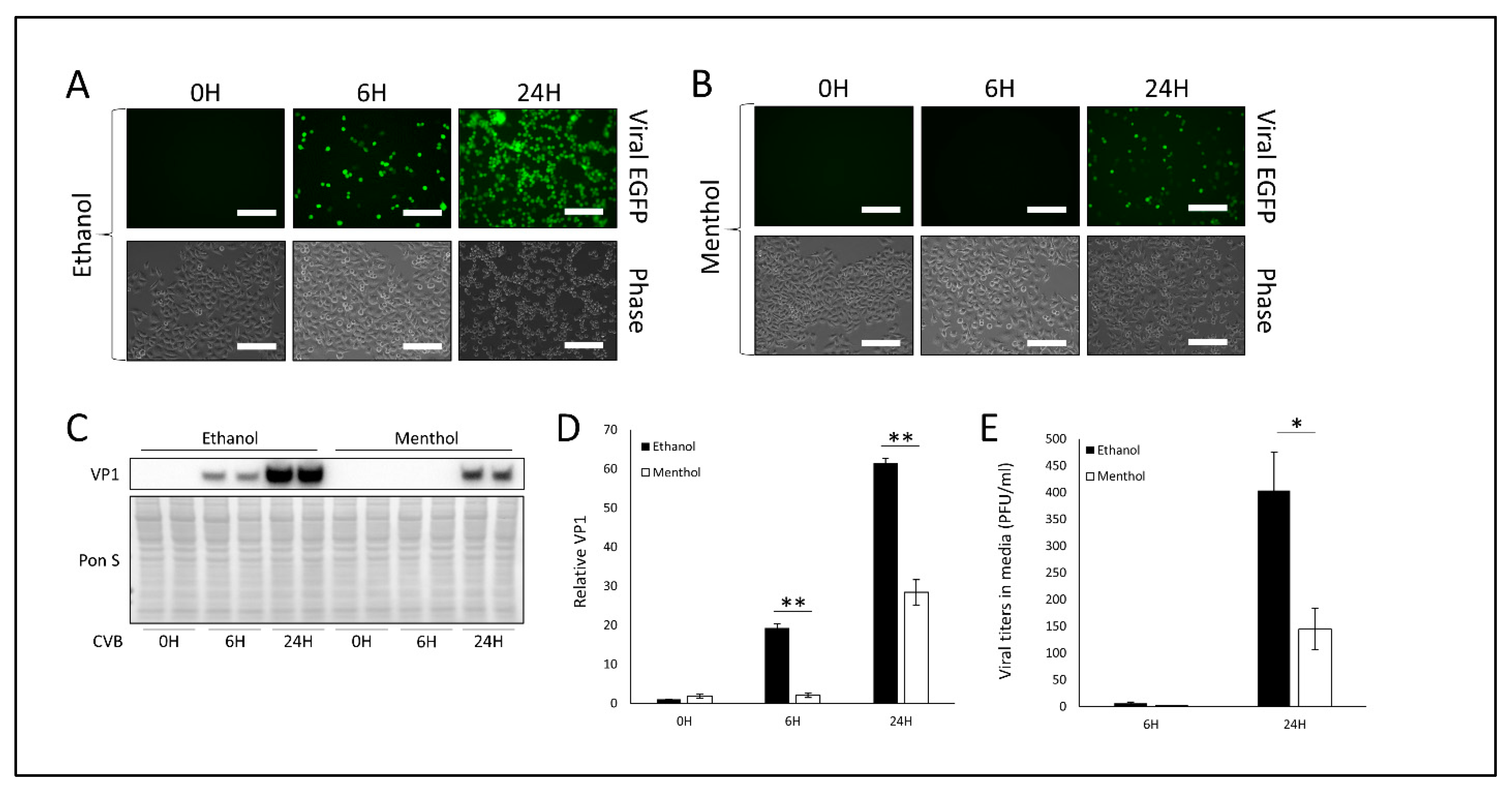
3.2. Treatment with TRPV1 Inhibitor SB-366791 or TRPM8 Agonist Menthol Attenuates CVB Infection
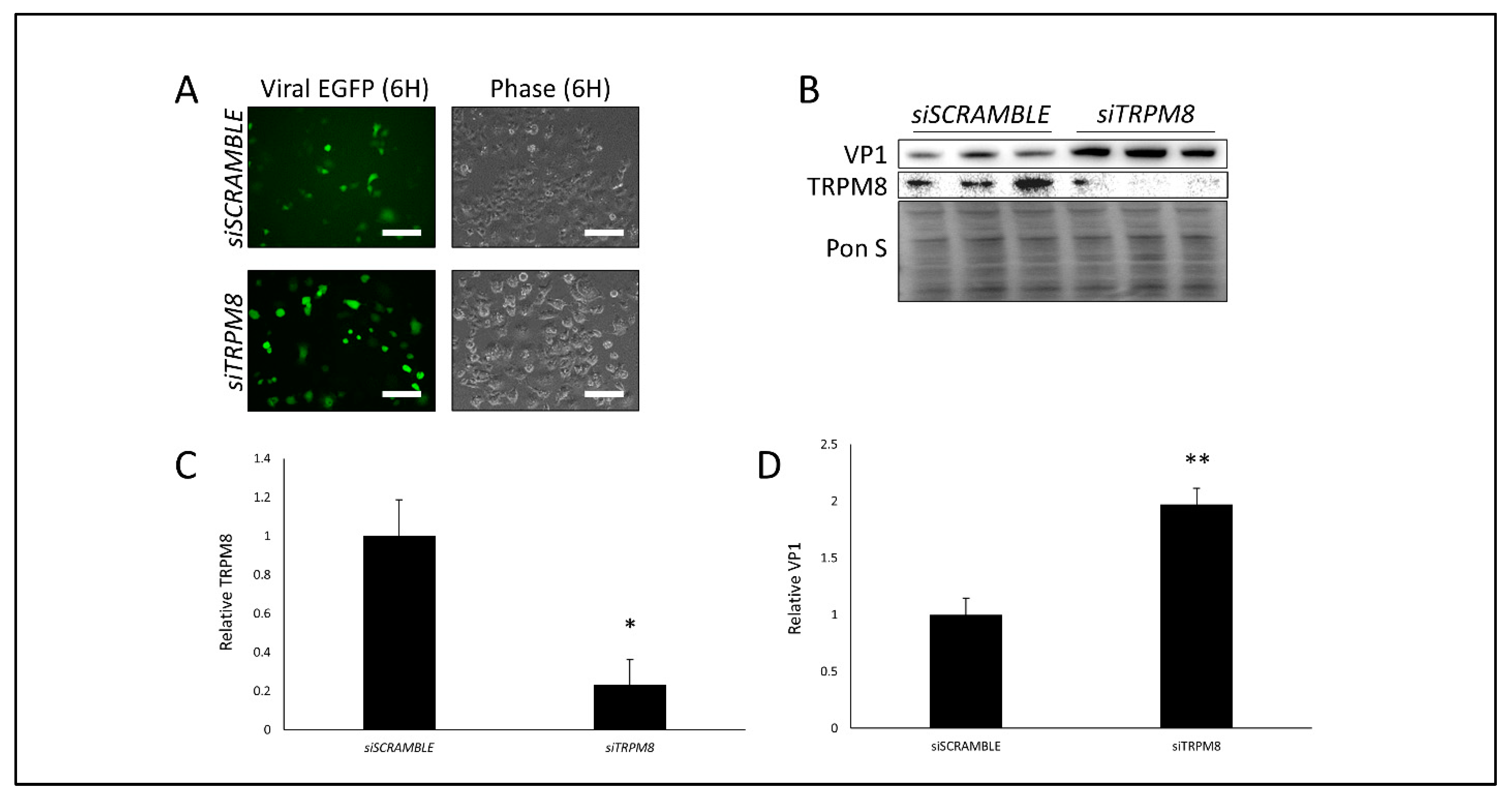
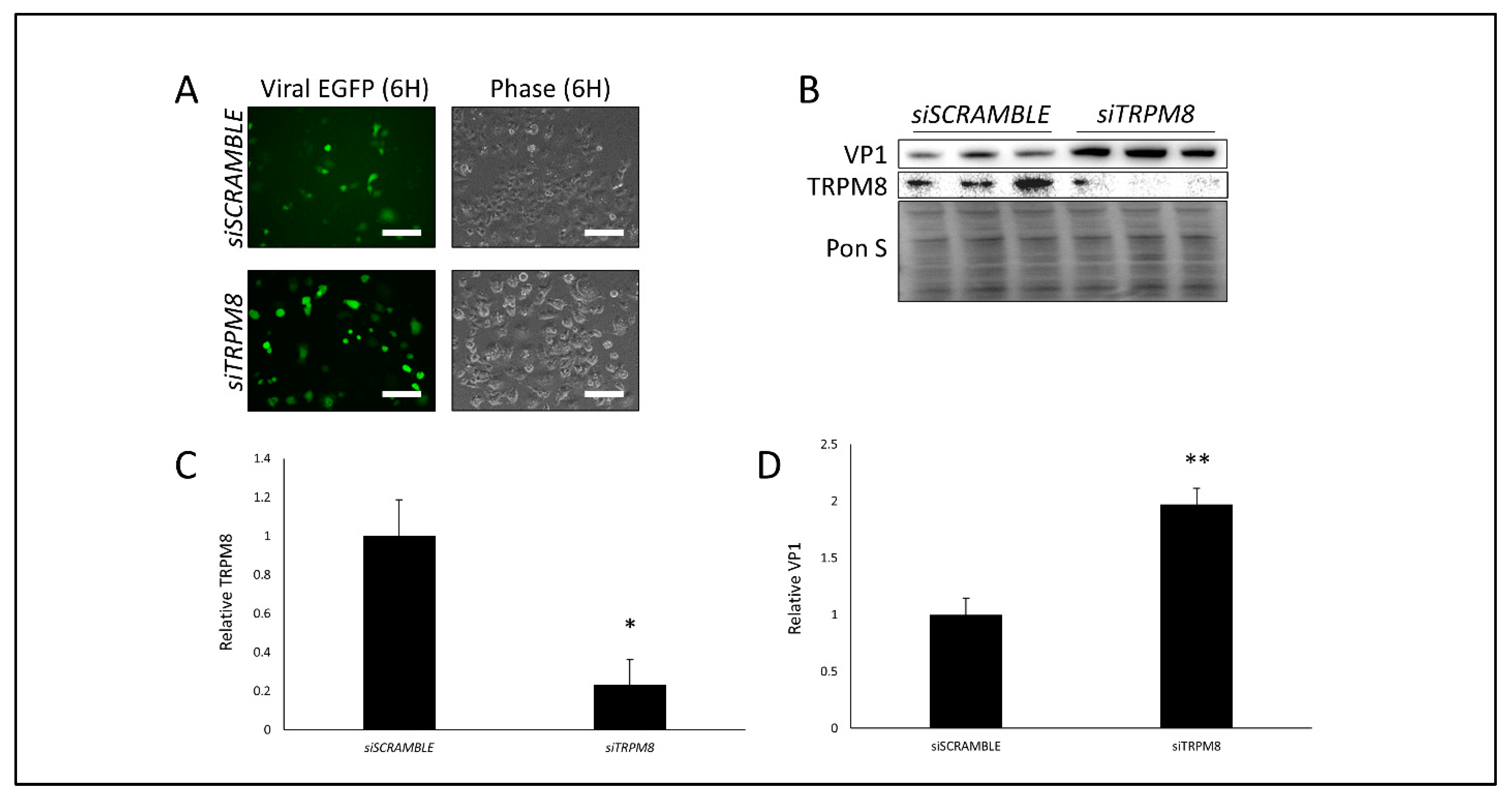
3.3. Silencing TRPM8 Bolsters CVB Infection
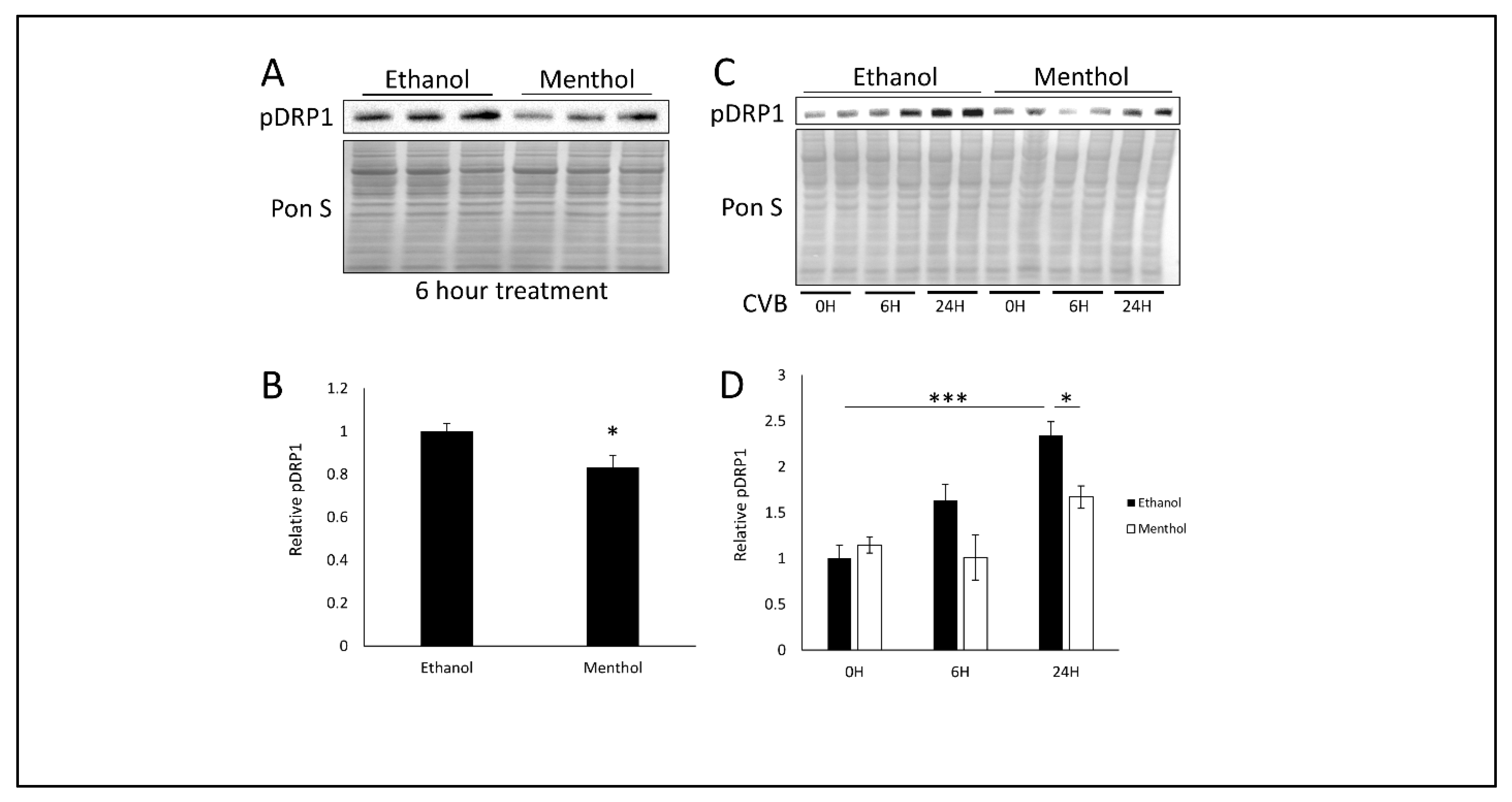
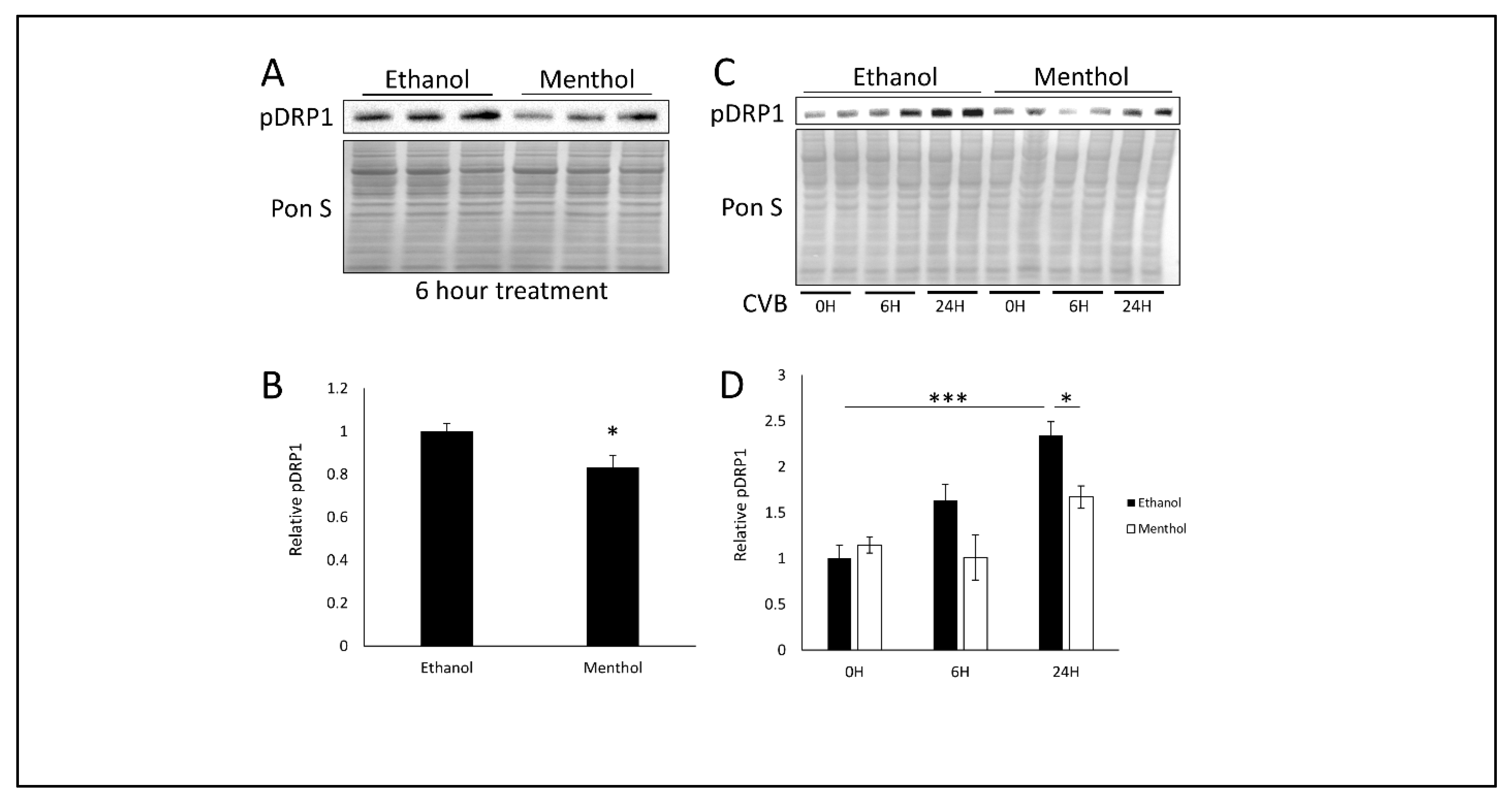
3.4. Menthol Treatment Reduces Mitochondrial Fission Basally and During CVB Infection
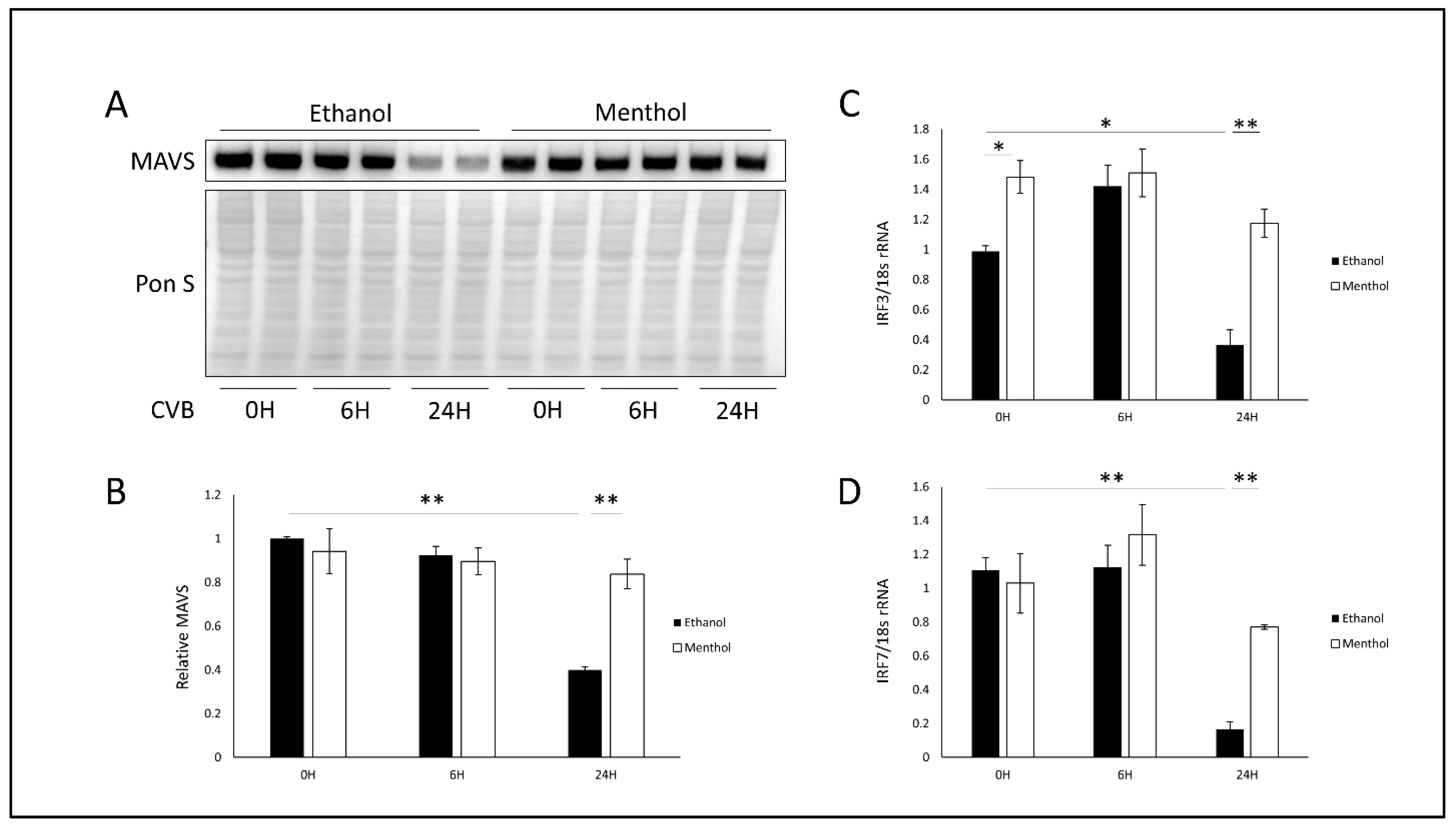
3.5. Menthol Enhances Antiviral Immunity During Infection
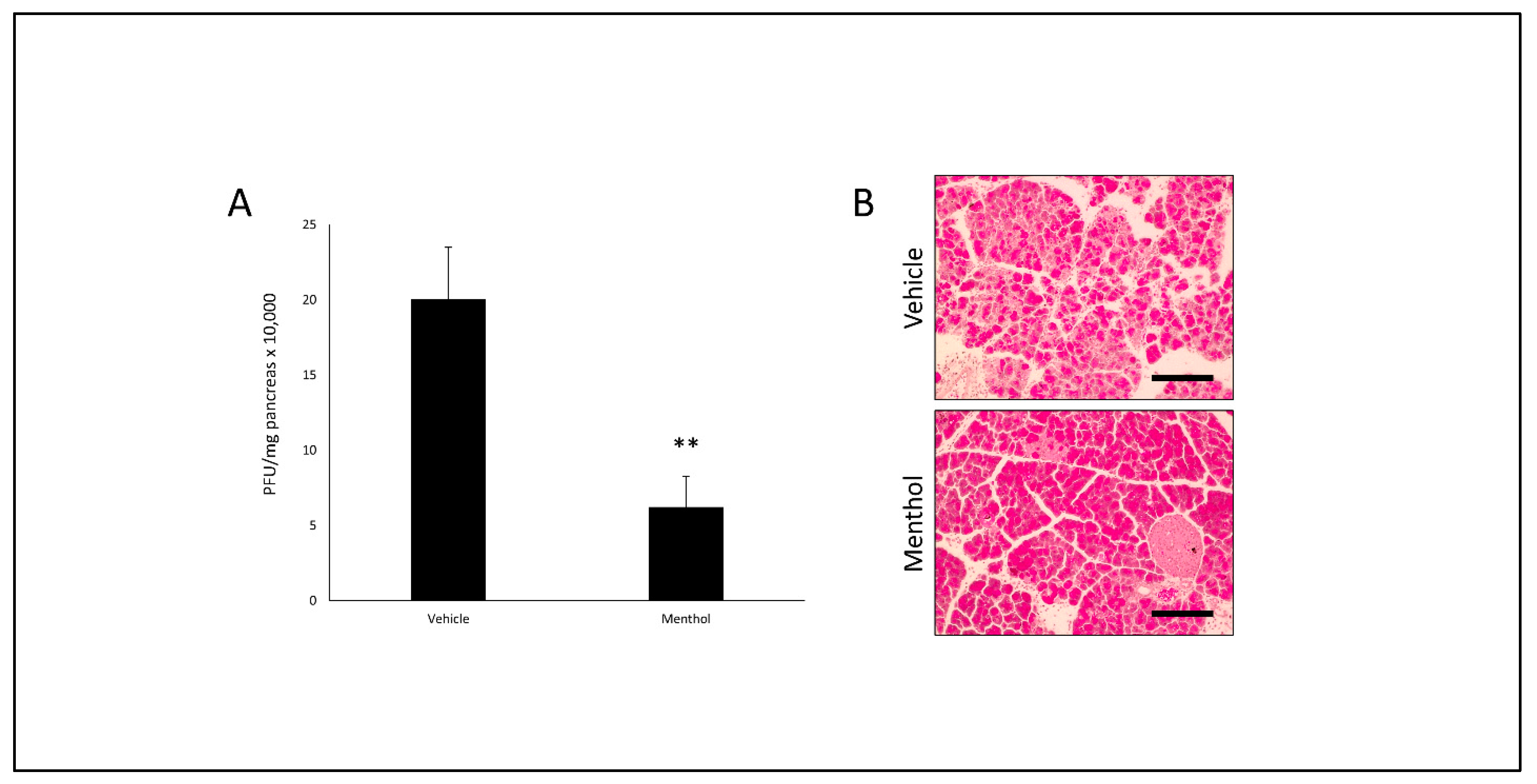
3.6. Oral Menthol Treatment Reduces Pancreatic CVB Titers and Tissue Destruction in CVB-Infected Mice
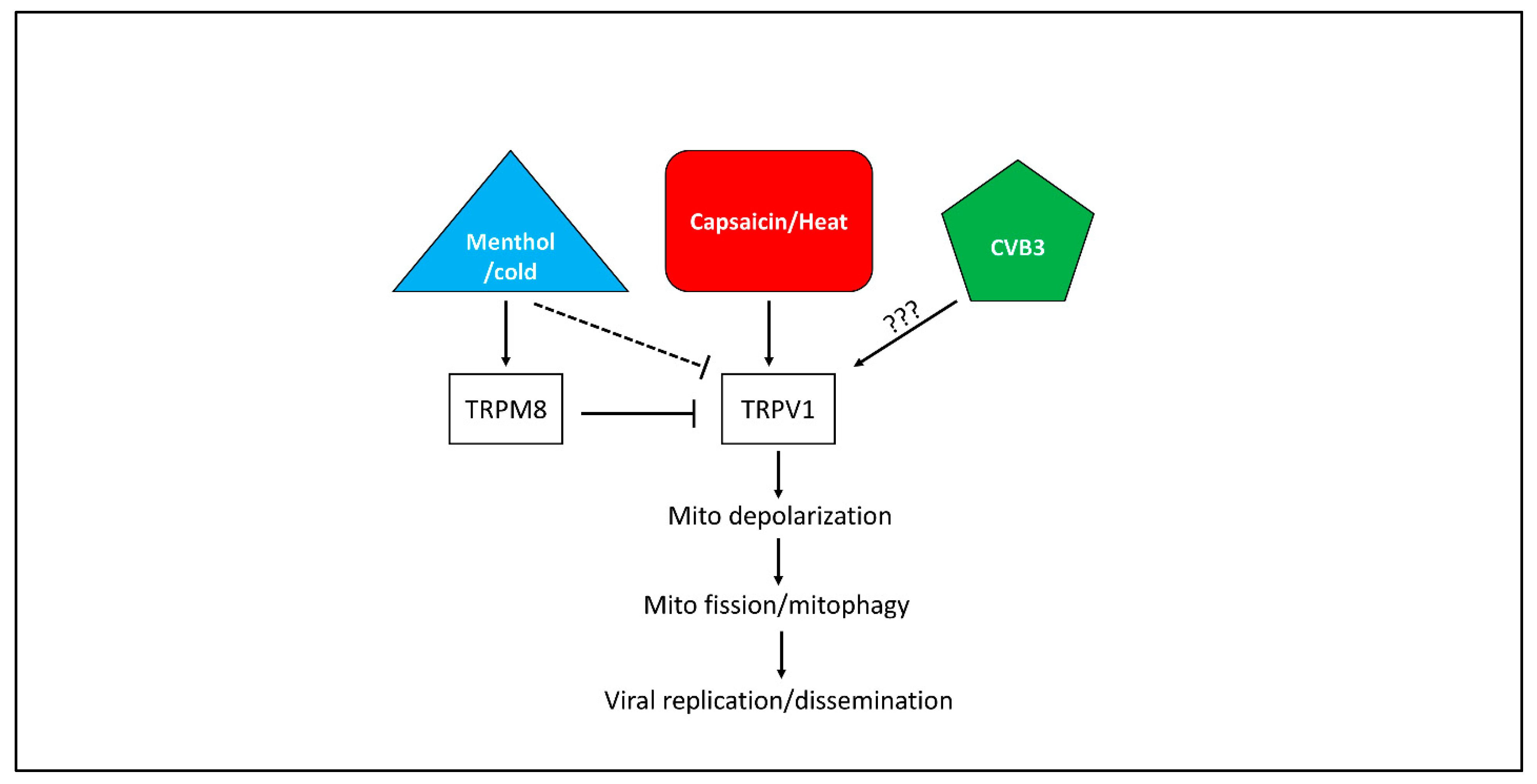
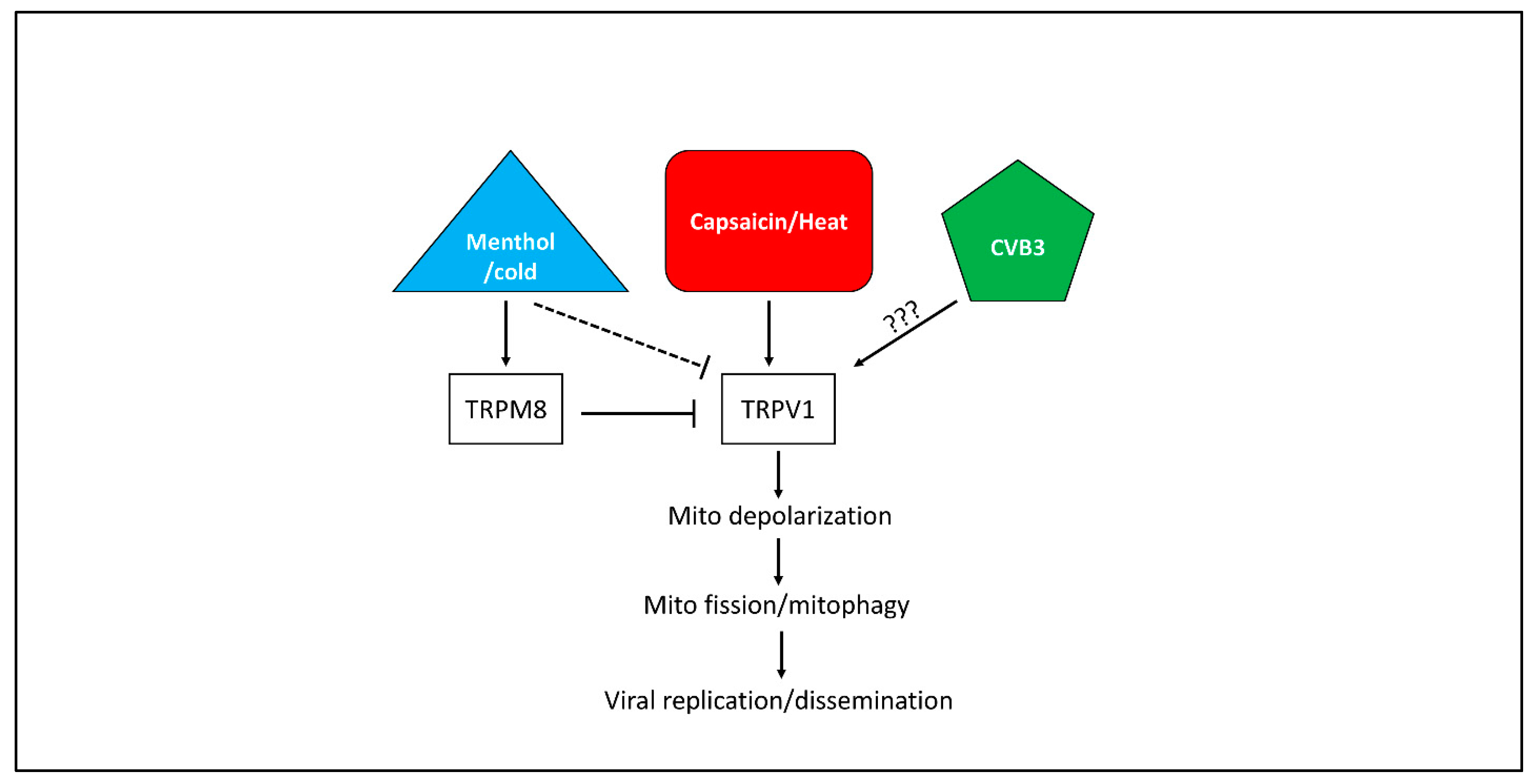
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tracy, S.; Hofling, K.; Pirruccello, S.; Lane, P.H.; Reyna, S.M.; Gauntt, C.J. Group B coxsackievirus myocarditis and pancreatitis: Connection between viral virulence phenotypes in mice. J. Med. Virol. 2000, 62, 70–81. [Google Scholar] [CrossRef]
- Huber, S.; Ramsingh, A.I. Coxsackievirus-induced pancreatitis. Viral Immunol. 2004, 17, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Hochman, D.; Louie, B.; Bailey, R. Determination of patient quality of life following severe acute pancreatitis. Can. J. Surg. 2006, 49, 101–106. [Google Scholar] [PubMed]
- Whitcomb, D.C. Clinical practice. Acute pancreatitis. N. Engl. J. Med. 2006, 354, 2142–2150. [Google Scholar] [CrossRef] [PubMed]
- Arnesjo, B.; Eden, T.; Ihse, I.; Nordenfelt, E.; Ursing, B. Enterovirus infections in acute pancreatitis—A possible etiological connection. Scand. J. Gastroenterol. 1976, 11, 645–649. [Google Scholar] [PubMed]
- Benifla, M.; Weizman, Z. Acute pancreatitis in childhood: Analysis of literature data. J. Clin. Gastroenterol. 2003, 37, 169–172. [Google Scholar] [CrossRef]
- Ostrowski, S.E.; Reilly, A.A.; Collins, D.N.; Ramsingh, A.I. Progression or resolution of coxsackievirus B4-induced pancreatitis: A genomic analysis. J. Virol. 2004, 78, 8229–8237. [Google Scholar] [CrossRef] [Green Version]
- Chrysos, G.; Kokkoris, S.; Protopsaltis, J.; Korantzopoulos, P.; Giannoulis, G. Coxsackievirus infection associated with acute pancreatitis. JOP 2004, 5, 384–387. [Google Scholar]
- Kemball, C.C.; Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Kiosses, W.B.; Whitton, J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 2010, 84, 12110–12124. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Sayen, M.R.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef]
- Luo, X.N.; Yao, H.L.; Song, J.; Song, Q.Q.; Shi, B.T.; Xia, D.; Jun, H.A.N. Coxsackievirus B3 Infection Triggers Autophagy through 3 Pathways of Endoplasmic Reticulum Stress. Biomed. Environ. Sci. 2018, 31, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.S.; Lin, S.C. AMPK Promotes Autophagy by Facilitating Mitochondrial Fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Yun, J.; Deng, H.; Guo, M. Atg1 mediated autophagy suppresses tissue degeneration in pink1/parkin mutants by promoting mitochondrial fission in Drosophila. Mol. Biol. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Zou, W.; Dai, X.; Yu, X.; Liu, H.; Chen, Q.; Teng, W. Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 2018, 25, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent roles of autophagy in virus infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [Green Version]
- Sin, J.; McIntyre, L.; Stotland, A.; Feuer, R.; Gottlieb, R.A. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.; Ohno, N.; Hsieh, Y.L.; Mahad, D.J.; Kikuchi, S.; Komuro, H.; Trapp, B.D.; Hsieh, S.-T. Mitochondrial fission augments capsaicin-induced axonal degeneration. Acta Neuropathol. 2015, 129, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Activation of mitochondrial transient receptor potential vanilloid 1 channel contributes to microglial migration. Glia 2015, 63, 1870–1882. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Barrantes, R.; Cordova, C.; Poblete, H.; Munoz, P.; Marchant, I.; Wianny, F.; Olivero, P. Perspectives of TRPV1 Function on the Neurogenesis and Neural Plasticity. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, M.; Uchida, K.; Suzuki, Y.; Matsui, H.; Shimada, T.; Fujita, F.; Tominaga, M. Reciprocal effects of capsaicin and menthol on thermosensation through regulated activities of TRPV1 and TRPM8. J. Physiol. Sci. 2016, 66, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Feuer, R.; Mena, I.; Pagarigan, R.; Slifka, M.K.; Whitton, J.L. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 2002, 76, 4430–4440. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.A.; Connor, M. TRPV1 antagonists as a potential treatment for hyperalgesia. Recent Pat. CNS Drug Discov. 2006, 1, 65–76. [Google Scholar] [CrossRef]
- Lee, L.Y.; Gu, Q. Role of TRPV1 in inflammation-induced airway hypersensitivity. Curr. Opin. Pharmacol. 2009, 9, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Zhang, M.; Yan, R.; Shan, H.; Diao, J.; Wei, J. Inhibition of Drp1 attenuates mitochondrial damage and myocardial injury in Coxsackievirus B3 induced myocarditis. Biochem. Biophys. Res. Commun. 2017, 484, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.S.; La, J.H.; Scheff, N.N.; Davis, B.M.; Albers, K.M.; Gebhart, G.F. TRPV1 and TRPA1 antagonists prevent the transition of acute to chronic inflammation and pain in chronic pancreatitis. J. Neurosci. 2013, 33, 5603–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, E.S.; Christianson, J.A.; Chen, X.; La, J.H.; Davis, B.M.; Albers, K.M.; Gebhart, G.F. Synergistic role of TRPV1 and TRPA1 in pancreatic pain and inflammation. Gastroenterology 2011, 140, 1283–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigna, S.R.; Shahid, R.A.; Liddle, R.A. Ethanol contributes to neurogenic pancreatitis by activation of TRPV1. FASEB J. 2014, 28, 891–896. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.J.; Lee, H.S. Experimental models of pancreatitis. Clin. Endosc. 2014, 47, 212–216. [Google Scholar] [CrossRef]
- Omar, S.; Clarke, R.; Abdullah, H.; Brady, C.; Corry, J.; Winter, H.; Cosby, S.L.; Touzelet, Q.; Power, U.F.; Lundy, F.; et al. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE 2017, 12, e0171681. [Google Scholar] [CrossRef] [Green Version]
- Schuhmacher, A.; Reichling, J.; Schnitzler, P. Virucidal effect of peppermint oil on the enveloped viruses herpes simplex virus type 1 and type 2 in vitro. Phytomedicine 2003, 10, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Germano, J.F.; Sawaged, S.; Saadaeijahromi, H.; Andres, A.M.; Feuer, R.; Gottlieb, R.A.; Sin, J. Coxsackievirus B infection induces the extracellular release of miR-590-5p, a proviral microRNA. Virology 2019, 529, 169–176. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, D.J.R.; Hamid, S.M.; Andres, A.M.; Saadaeijahromi, H.; Piplani, H.; Germano, J.F.; Song, Y.; Sawaged, S.; Feuer, R.; Pandol, S.J.; et al. Antiviral Effects of Menthol on Coxsackievirus B. Viruses 2020, 12, 373. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040373
Taylor DJR, Hamid SM, Andres AM, Saadaeijahromi H, Piplani H, Germano JF, Song Y, Sawaged S, Feuer R, Pandol SJ, et al. Antiviral Effects of Menthol on Coxsackievirus B. Viruses. 2020; 12(4):373. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040373
Chicago/Turabian StyleTaylor, David J.R., Syed M. Hamid, Allen M. Andres, Hannaneh Saadaeijahromi, Honit Piplani, Juliana F. Germano, Yang Song, Savannah Sawaged, Ralph Feuer, Stephen J. Pandol, and et al. 2020. "Antiviral Effects of Menthol on Coxsackievirus B" Viruses 12, no. 4: 373. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040373