Genomic Analyses of Potential Novel Recombinant Human Adenovirus C in Brazil

 ,
,  , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Viral Metagenomics
2.3. Phylogenetic Analysis
2.4. Detection of Recombination
2.5. Homology Modeling of Hexon Structure
2.6. Ethical Approval
3. Results
3.1. Genetic Distances of Brazilian HAdV-C
3.2. Phylogenetic Analysis of Near Full-Length Genomes of HAdV-C
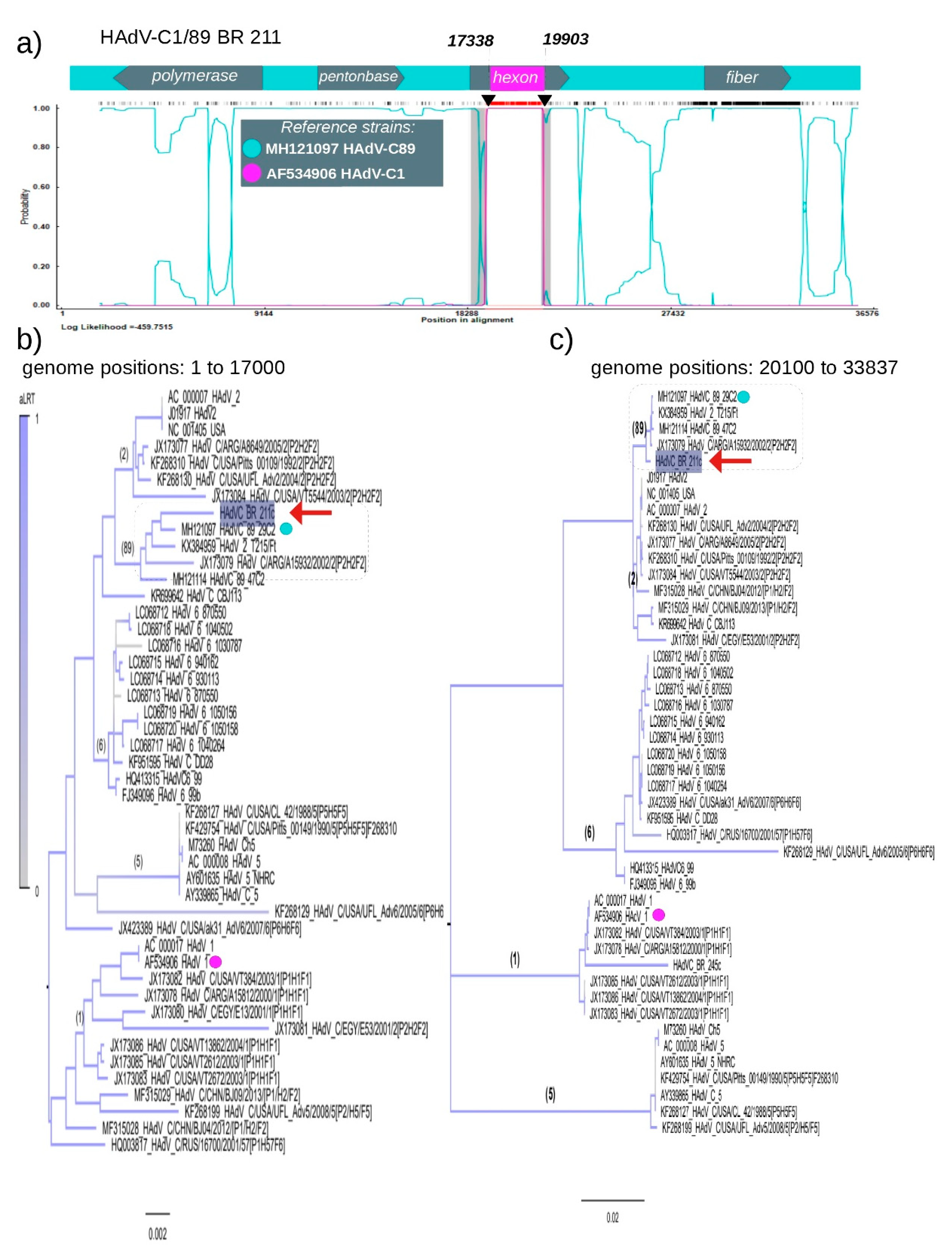
3.3. Recombination of HAdV-C1 and HAdV-C89
3.4. Structure of Recombinant Hexon Protein of HAdV-C
3.5. Characteristics of Penton Base of HAdV-C BR-211
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rivailler, P.; Mao, N.; Zhu, Z.; Xu, W. Recombination analysis of human mastadenovirus c whole genomes. Sci. Rep. 2019, 9, 2182. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular evolution of human adenoviruses. Sci. Rep. 2013, 3, 1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratte-Santos, R.; Miagostovich, M.P.; Fumian, T.M.; Maciel, E.L.; Martins, S.A.; Cassini, S.T.; Keller, R. High prevalence of enteric viruses associated with acute gastroenteritis in pediatric patients in a low-income area in vitoria, southeastern brazil. J. Med. Virol. 2019, 91, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Filho, E.P.; da Costa Faria, N.R.; Fialho, A.M.; de Assis, R.S.; Almeida, M.M.S.; Rocha, M.; Galvao, M.; Dos Santos, F.B.; Barreto, M.L.; Leite, J.P.G. Adenoviruses associated with acute gastroenteritis in hospitalized and community children up to 5 years old in rio de janeiro and salvador, brazil. J. Med. Microbiol. 2007, 56, 313–319. [Google Scholar] [CrossRef]
- Wang, Y.F.; Shen, F.C.; Wang, S.L.; Kuo, P.H.; Tsai, H.P.; Liu, C.C.; Wang, J.R.; Chi, C.Y. Molecular epidemiology and clinical manifestations of adenovirus respiratory infections in taiwanese children. Medicine (Baltimore) 2016, 95, e3577. [Google Scholar] [CrossRef]
- Dhingra, A.; Hage, E.; Ganzenmueller, T.; Bottcher, S.; Hofmann, J.; Hamprecht, K.; Obermeier, P.; Rath, B.; Hausmann, F.; Dobner, T.; et al. Molecular evolution of human adenovirus (hadv) species c. Sci. Rep. 2019, 9, 1039. [Google Scholar] [CrossRef]
- Yang, J.; Mao, N.; Zhang, C.; Ren, B.; Li, H.; Li, N.; Chen, J.; Zhang, R.; Li, H.; Zhu, Z.; et al. Human adenovirus species c recombinant virus continuously circulated in china. Sci. Rep. 2019, 9, 9781. [Google Scholar] [CrossRef]
- Alonso-Padilla, J.; Papp, T.; Kajan, G.L.; Benko, M.; Havenga, M.; Lemckert, A.; Harrach, B.; Baker, A.H. Development of novel adenoviral vectors to overcome challenges observed with hadv-5-based constructs. Mol. Ther. 2016, 24, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.P.; Seto, J.; Liu, E.B.; Dehghan, S.; Hudson, N.R.; Lukashev, A.N.; Ivanova, O.; Chodosh, J.; Dyer, D.W.; Jones, M.S.; et al. Computational analysis of two species c human adenoviruses provides evidence of a novel virus. J. Clin. Microbiol. 2011, 49, 3482–3490. [Google Scholar] [CrossRef] [Green Version]
- Houldcroft, C.J.; Roy, S.; Morfopoulou, S.; Margetts, B.K.; Depledge, D.P.; Cudini, J.; Shah, D.; Brown, J.R.; Romero, E.Y.; Williams, R.; et al. Use of whole-genome sequencing of adenovirus in immunocompromised pediatric patients to identify nosocomial transmission and mixed-genotype infection. J. Infect. Dis. 2018, 218, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Mao, N.; Zhu, Z.; Rivailler, P.; Chen, M.; Fan, Q.; Huang, F.; Xu, W. Whole genomic analysis of two potential recombinant strains within human mastadenovirus species c previously found in beijing, china. Sci. Rep. 2017, 7, 15380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlys da Costa, A.; Theze, J.; Komninakis, S.C.V.; Sanz-Duro, R.L.; Felinto, M.R.L.; Moura, L.C.C.; Barroso, I.M.O.; Santos, L.E.C.; Nunes, M.A.L.; Moura, A.A.; et al. Spread of chikungunya virus east/central/south african genotype in northeast brazil. Emerg. Infect. Dis. 2017, 23, 1742–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilli, A.; Luchs, A.; Leal, E.; Gill, D.; Milagres, F.A.P.; Komninakis, S.V.; Brustulin, R.; Teles, M.; Lobato, M.; Chagas, R.T.D.; et al. Human sapovirus gi.2 and gi.3 from children with acute gastroenteritis in northern brazil. Memórias Inst. Oswaldo Cruz 2019, 114, e180574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Costa, A.C.; Leal, E.; Gill, D.; Milagres, F.A.P.; Komninakis, S.V.; Brustulin, R.; Teles, M.; Lobato, M.; das Chagas, R.T.; Abrao, M.; et al. Discovery of cucumis melo endornavirus by deep sequencing of human stool samples in brazil. Virus Genes 2019, 55, 332–338. [Google Scholar] [CrossRef]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Posada, D. Jmodeltest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. Mega x: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The swiss-model workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Soding, J. Hhblits: Lightning-fast iterative protein sequence searching by hmm-hmm alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.C.; Luchs, A.; Milagres, F.A.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.; Brustulin, R.; das Chagas, R.T.; Abrao, M.; Soares, C.; et al. Recombination located over 2a-2b junction ribosome frameshifting region of saffold cardiovirus. Viruses 2018, 10, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubieta, C.; Blanchoin, L.; Cusack, S. Structural and biochemical characterization of a human adenovirus 2/12 penton base chimera. FEBS J. 2006, 273, 4336–4345. [Google Scholar] [CrossRef]
- da Costa, A.C.; Luchs, A.; Milagres, F.A.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.; Brustulin, R.; das Chagas, R.T.; Abrao, M.; Soares, C.; et al. Near full length genome of a recombinant (e/d) cosavirus strain from a rural area in the central region of brazil. Sci. Rep. 2018, 8, 12304. [Google Scholar] [CrossRef]
- Leal, E.; Luchs, A.; Milagres, F.A.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.; Brustulin, R.; Chagas, R.T.D.; Abrao, M.; Soares, C.; et al. Recombinant strains of human parechovirus in rural areas in the north of brazil. Viruses 2019, 11, 488. [Google Scholar] [CrossRef] [Green Version]
- Luchs, A.; Leal, E.; Komninakis, S.V.; de Padua Milagres, F.A.; Brustulin, R.; da Aparecida Rodrigues Teles, M.; Gill, D.E.; Deng, X.; Delwart, E.; Sabino, E.C.; et al. Wuhan large pig roundworm virus identified in human feces in brazil. Virus Genes 2018, 54, 470–473. [Google Scholar] [CrossRef]
- Luchs, A.; Leal, E.; Tardy, K.; Milagres, F.A.P.; Komninakis, S.V.; Brustulin, R.; Teles, M.; Lobato, M.; das Chagas, R.T.; Abrao, M.; et al. The rare enterovirus c99 and echovirus 29 strains in brazil: Potential risks associated to silent circulation. Memórias Inst. Oswaldo Cruz 2019, 114, e190160. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, G.O.; Luchs, A.; Milagres, F.A.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.; Brustulin, R.; Chagas, R.T.D.; Abrao, M.; Soares, C.; et al. Detection and characterization of enterovirus b73 from a child in brazil. Viruses 2018, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Rosa, U.A.; Ribeiro, G.O.; Villanova, F.; Luchs, A.; Milagres, F.A.P.; Komninakis, S.V.; Tahmasebi, R.; Lobato, M.; Brustulin, R.; Chagas, R.T.D.; et al. First identification of mammalian orthoreovirus type 3 by gut virome analysis in diarrheic child in brazil. Sci. Rep. 2019, 9, 18599. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.S.A.; Luchs, A.; Leal, E.; Milagres, F.A.P.; Komninakis, S.V.; Gill, D.E.; Lobato, M.; Brustulin, R.; das Chagas, R.T.; Abrao, M.; et al. Complete genome sequences of six human bocavirus strains from patients with acute gastroenteritis in the north region of brazil. Genome Announc. 2018, 6, e00235-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afrad, M.H.; Avzun, T.; Haque, J.; Haque, W.; Hossain, M.E.; Rahman, A.R.; Ahmed, S.; Faruque, A.S.G.; Rahman, M.Z.; Rahman, M. Detection of enteric- and non-enteric adenoviruses in gastroenteritis patients, bangladesh, 2012–2015. J. Med. Virol. 2018, 90, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Ghebremedhin, B. Human adenovirus: Viral pathogen with increasing importance. Eur. J. Microbiol. Immunol. (Bp.) 2014, 4, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Primo, D.; Pacheco, G.T.; Timenetsky, M.; Luchs, A. Surveillance and molecular characterization of human adenovirus in patients with acute gastroenteritis in the era of rotavirus vaccine, brazil, 2012–2017. J. Clin. Virol. 2018, 109, 35–40. [Google Scholar] [CrossRef]
- Ismail, A.M.; Zhou, X.; Dyer, D.W.; Seto, D.; Rajaiya, J.; Chodosh, J. Genomic foundations of evolution and ocular pathogenesis in human adenovirus species d. FEBS Lett. 2019, 593, 3583–3608. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Sun, X.; Ye, X.; Feng, Y.; Wang, J.; Zheng, X.; Liu, X.; Yi, C.; Hao, M.; Wang, Q.; et al. Hexon and fiber of adenovirus type 14 and 55 are major targets of neutralizing antibody but only fiber-specific antibody contributes to cross-neutralizing activity. Virology 2018, 518, 272–283. [Google Scholar] [CrossRef]
- Liu, T.; Fan, Y.; Li, X.; Gu, S.; Zhou, Z.; Xu, D.; Qiu, S.; Li, C.; Zhou, R.; Tian, X. Identification of adenovirus neutralizing antigens using capsid chimeric viruses. Virus Res. 2018, 256, 100–106. [Google Scholar] [CrossRef]
- Tian, X.; Qiu, H.; Zhou, Z.; Wang, S.; Fan, Y.; Li, X.; Chu, R.; Li, H.; Zhou, R.; Wang, H. Identification of a critical and conformational neutralizing epitope in human adenovirus type 4 hexon. J. Virol. 2018, 92, e01643-17. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Region | Type 1 | Type 2 | Type 5 | Type 6 | Type 57 | Type 89 | Strain |
|---|---|---|---|---|---|---|---|
| Penton base (P) | 0.00886 | 0.01065 | 0.01424 | 0.00886 | 0.00896 | 0.00510 | BR-211 |
| 0.00235 | 0.00886 | 0.01663 | 0.00471 | 0.00335 | 0.01543 | BR-245 | |
| Hexon (H) | 0.00242 | 0.16695 | 0.20007 | 0.19355 | 0.13400 | 0.17017 | BR-211 |
| 0.00446 | 0.017060 | 0.20023 | 0.19549 | 0.14343 | 0.17385 | BR-245 | |
| Fiber (F) | 0.777510 | 0.004093 | 0.722381 | 0.685276 | 0.692742 | 0.000994 | BR-211 |
| 0.025583 | 0.775442 | 0.691595 | 0.598394 | 0.605335 | 0.779578 | BR-245 |
| Penton Base Position | HAdV-C | ||||
|---|---|---|---|---|---|
| BR-211 | Type 89 | Type 2 | Type 5 | Type 1 | |
| 2 | R | R | Q | R | R |
| 153 | Q | Q | L | P | L |
| 157 | K | N | K | N | K |
| 312 | S | S | N | S | N |
| 458 | R | R | S | R | R/S |
| 361–364 | AAAP | Del. | AAAP | AAAP | AAAP |
| 367–369 | EAA | EAA | Del. | Del. | EAA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahmasebi, R.; da Costa, A.C.; Tardy, K.; J. Tinker, R.; de Padua Milagres, F.A.; Brustulin, R.; Rodrigues Teles, M.d.A.; Togisaki das Chagas, R.; de Deus Alves Soares, C.V.; Sakurada Aranha Watanabe, A.; et al. Genomic Analyses of Potential Novel Recombinant Human Adenovirus C in Brazil. Viruses 2020, 12, 508. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050508
Tahmasebi R, da Costa AC, Tardy K, J. Tinker R, de Padua Milagres FA, Brustulin R, Rodrigues Teles MdA, Togisaki das Chagas R, de Deus Alves Soares CV, Sakurada Aranha Watanabe A, et al. Genomic Analyses of Potential Novel Recombinant Human Adenovirus C in Brazil. Viruses. 2020; 12(5):508. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050508
Chicago/Turabian StyleTahmasebi, Roozbeh, Antonio Charlys da Costa, Kaelan Tardy, Rory J. Tinker, Flavio Augusto de Padua Milagres, Rafael Brustulin, Maria da Aparecida Rodrigues Teles, Rogério Togisaki das Chagas, Cassia Vitória de Deus Alves Soares, Aripuana Sakurada Aranha Watanabe, and et al. 2020. "Genomic Analyses of Potential Novel Recombinant Human Adenovirus C in Brazil" Viruses 12, no. 5: 508. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050508