Genome Analysis of a Novel Tembusu Virus in Taiwan


Center for Diagnostics and Vaccine Development, Centers for Disease Control, Ministry of Health and Welfare, Taipei City 11561, Taiwan
*
Author to whom correspondence should be addressed.
Viruses 2020, 12(5), 567; https://0-doi-org.brum.beds.ac.uk/10.3390/v12050567
Submission received: 7 April 2020
/
Revised: 20 May 2020
/
Accepted: 20 May 2020
/
Published: 22 May 2020
(This article belongs to the Section Animal Viruses)
Abstract
:We identified and isolated a novel Tembusu virus (TMUV) strain TP1906 (TMUV-TP1906) from a Culex annulus mosquito pool collected from the northern part of Taiwan in 2019. The TMUV-TP1906 genome is a 10,990-nucleotide-long, positive-sense, single-stranded RNA, consisting of a single open reading frame (ORF) encoding a polyprotein of 3425 amino acids, with 5′ and 3′ untranslated regions (UTRs) of 94 and 618 nucleotides, respectively. The nucleotide sequence of the TMUV-TP1906 of ORF exhibited 93.71% and 91.27% similarity with Sitiawan virus (STWV) and the TMUV prototype strain MM1775, respectively. The 3′-UTR variable region of TMUV-TP1906 showed nucleotide sequence divergence with other TMUV strains. Phylogenetic analysis of the complete ORF and polyprotein sequences revealed that TMUV-TP1906 is most closely related to STWV which causes encephalitis and retarded growth in chickens. We found that the TMUV-TP1906 caused a cytopathic effect (CPE) in the DF-1 chicken fibroblast cell line, while no apparent CPE was observed in Vero and C6/36 cells. In this study, we first identified and isolated a novel TMUV strain in Taiwan. In addition, to our knowledge, it is the first time that the TMUV strain was isolated from the Cx. annulus mosquitoes. Further study is warranted to investigate the host range and virulence of TMUV-TP1906.
1. Introduction
Tembusu virus (TMUV) is a single-stranded, positive-sense RNA virus that belongs to the Flaviviridae family, Flavivirus genus and Ntaya virus group. The TMUV prototype strain MM1775 (TMUV-MM1775) was first isolated from Culex tritaeniorhynchus mosquitoes in Kuala Lumpur, Malaysia in 1955. Since 2000, many TMUV strains (TMUVs) have been isolated from birds and mosquitoes, including chicks, ducks, geese, pigeons, sparrows and Culex mosquitoes. TMUV consists of several genetically closely related virus strains with noticeable pathogenicity for poultry, such as Sitiawan virus (STWV), duck TMUV (DTMUV), Perak virus and Baiyangdian virus [1]. STWV was isolated from sick broiler chicks in Sitiawan District of Perak State, Malaysia in 2000 [2]. STWV-infected chicks showed encephalitis, growth retardation and increased blood sugar levels. In 2010, DTMUV, also called duck egg-drop syndrome virus, was found to extensively infect ducks with high morbidity (up to 90%) and mortality (5% to 30%) rates in southeast China [3,4]. DTMUV outbreaks also occurred in layer and broiler duck farms in Pekin ducks in Malaysia in 2012 and in Thailand in 2013 [5,6].
The TMUV genome is approximately 11,000 nucleotides and contains 5′ and 3′ untranslated regions (UTRs), and a long open reading frame (ORF) that encodes three structural proteins (capsid (C), pre-membrane protein (prM) and envelope (E)) and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). Phylogenetic analysis of the nucleotide sequences of the ORFs of several TMUVs showed that TMUV-MM1775 and STWV clustered together away from the genomes of other DTMUVs [7].
TMUVs have been isolated from several mosquito species in Asia, mainly Cx. tritaeniorhynchus [8,9] and also Culex species, including Cx. vishnui, Cx. gelidus and Cx. pipiens [5,9,10]. A mosquito vector competence study by O’Guinn et al. demonstrated that Cx. vishnui developed high viral titers after feeding on TMUV-infected chicks and could readily transmit TMUV to naive chickens [11]. Although TMUV are considered mosquito-borne viruses [4], in vivo studies suggested that TMUVs could be transmitted among ducks by direct contact and aerosol transmission [12,13]. The role of mosquitoes as vectors in the transmission of TMUV to avian hosts still needs further study.
Taiwan is an island off the southeastern coast of mainland China that is near the epicenter of DTMUV outbreaks. Taiwan straddles the Tropic of Cancer, having a warm tropical-subtropical climate and large variety of mosquito species and is also an important rest point for migrating birds [14]. To monitor arboviruses in Taiwan, we conducted a survey on mosquitoes collected from wetlands, pig farms and parks in Taiwan between April and September 2019. In this study, we identified novel TMUV strains in Cx. annulus and Cx. tritaeniorhynchus mosquitoes collected from northern and central parts of Taiwan, respectively. We isolated and characterized the genetic sequence of this novel virus.
2. Materials and Methods
2.1. Collections of Mosquitoes
The mosquitoes were collected on pig farms near rice paddy fields in the northern (Wujie Township in Yilan County), central (Wufeng District in Taichung City), southern (Xiaying District in Tainan City), and eastern (Shoufeng Townships in Hualien County) regions; from a wetland habitat for waterbirds in the northern (Beitou District in Taipei City) region; and also from community parks in the northern (Shilin District in Taipei City and Luodong Township in Yilan County), central (Nantun District in Taichung City) and southern (North District in Tainan City) regions of Taiwan between April and September, 2019 (Figure 1). The mosquitoes were collected using dry ice traps in wetland and parks and sweep nets in pig farm and were transported either alive or on dry ice to the laboratory. Mosquito collections by sweep nets were conducted only on the same day between 18:30 and 20:30 in the pig farms, while dry ice traps were set up overnight from 17:00 to 8:00 the next morning in wetland and parks as previously described [15]. Only female mosquitoes were analyzed in this study. The mosquitoes were frozen at –20 °C and further examined and classified according to the characteristics of the species. The pooled (1 to 50) mosquitoes were used for RNA extraction and virus isolation. The mosquito pools were homogenized in a TissueLyzer (Qiagen GmbH, Hilden, Germany) with two cycles at 4 °C for 90 sec at a frequency of 30 Hz after adding a 3 mm steel ball to each tube and 0.6 mL RPMI 1640 liquid medium. The pools were then clarified by centrifugation. The supernatants were sterilized by filtration and removed for RNA extraction and virus isolation.
2.2. RNA Extraction and Real Time RT-PCR
Viral RNA was extracted from the mosquito suspensions or cell culture supernatants using the QIAamp viral RNA mini kit (Qiagen GmbH, Hilden, Germany). The flavivirus-specific primers (1370F: 5′-TGY GTB TAC AAC ATG ATG GG; 1442F: 5′-ATA TGG TAC ATG TGG CTA GGA GC and 1620R: 5′-GTG TCC CAN CCH GCT GTG TCA) were mixed and used for the RT-PCR screening assay. Real-time RT-PCR was performed using a QuantiTect SYBR Green RT-PCR kit (Qiagen GmbH, Hilden, Germany) with the following parameters: 50 °C for 30 min; 95 °C for 15 min and 45 cycles of 95 °C for 15 s, 55 °C for 30 s, 72 °C for 20 s, and 77 °C for 20 s; and 95 °C for 1 min, melting curve program from 68 °C to 95 °C [16,17]. DNA sequencing of positive RT-PCR products was performed, and RT-PCR positive mosquito pools were subjected to virus isolation.
2.3. Virus Isolation and Immunofluorescence Assay (IFA)
Filtered homogenates of mosquito pools were initially cultured with the Aedes albopictus C6/36 cell line or Vero cell line. C6/36 cells were infected with TMUV-TP1906 and tested by IFA. Briefly, cells were fixed and incubated with anti-flavivirus monoclonal antibody (D56.3) for 1 h. The cells were subsequently washed and incubated with FITC-conjugated goat anti-mouse IgG antibody (Invitrogen, Carlsbab, CA, USA) for 1 h. After washing, the images of the cells were examined using a fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
2.4. Viral Growth Kinetics
C6/36, Vero, BHK-21 and DF-1 cell lines were obtained from the American Type Culture Collection (ATCC; www.atcc.org) and used for the viral growth kinetics assays. C6/36 cells were grown in RPMI1640 medium supplemented with 5% fetal bovine serum (FBS) (Gibco, Invitrogen, Carlsbab, CA, USA), 100 units/mL–0.1 mg/mL–0.25 μg/mL penicillin-streptomycin-amphotericin B (PSA) (Gibco, Invitrogen), 1X non-essential amino acids (NEAA) (Gibco, Invitrogen) and incubated in 5% CO2 at 30 °C. Vero cells were grown in M199 medium supplemented with 5% FBS-PSA-NEAA and incubated in 5% CO2 at 37 °C. BHK-21 cells were cultured in MEM medium supplemented with 4% FBS-PSA-NEAA and incubated in 5% CO2 at 37 °C. DF-1 cells were cultured in DMEM medium supplemented with 10% FBS-PSA in 5% CO2 at 39 °C. Growth curves were performed by inoculating 0.1 mL TMUV-TP1906 (3.12 × 108 viral copies) into 80% confluent cells (5 × 105 cells per well) in 6-well plates (Corning, NY, USA). After 1 h incubation, medium with virus was removed and added 3 mL of fresh medium. All cell culture supernatants were collected at 0, 6, 24, 48, 72, 96 and 120 h post infection and the cell cultures were observed daily for cytopathic effect under a light microscopy (Carl Zeiss, Oberkochen, Germany). The viral RNA extracted from the supernatants was quantified by real time RT-PCR with the flavivirus-specific primer set and the viral copy number calculated according to the standard curve. All experiments were performed in triplicate.
2.5. Complete Genome Sequencing
RNA extracted from the cell culture supernatants and pellets from the first cell culture passage were used for RT-PCR and DNA sequencing. RT-PCR was performed using the Superscript III One-Step RT-PCR system with Platinum Taq High Fidelity (Invitrogen, Carlsbab, CA, USA). The RT-PCR parameters were as follows: 55 °C for 30 min; 94 °C for 2 min and 40 cycles of 94 °C for 15 s, 50 °C or 54 °C for 30 s, and 68 °C for 1 min; and a prolonged final extension at 68 °C for 5 min. The cDNA of the 5′ end of the viral genome was generated by rapid amplification of cDNA ends (RACE) using a 5′/3′ RACE kit, 2nd generation (Roche, Mannheim, Germany) with a gene specific primer (GSP-5′ end, Table 1). For the 3′ end, polyadenylated (polyA) tails were added to the 3′ ends of virus genomic ssRNA using T4 RNA ligase. Tailing reactions were performed at 37 °C for 1 h and used for RT-PCR directly. The 5′ and 3′ end fragments were amplified using the genome-specific primer GSP-5′ end and 3′UTR-10 with oligo d(T)-anchor primer (provided by the 5′/3′ RACE kit), respectively (listed in Table 1). The PCR products were sequenced directly by using the BigDye Terminator Cycle Sequencing Kit and the ABI 3730xl DNA analyzer (Applied Biosystems) according to the manufacturer’s protocols. Each forward and reverse primer was used for sequencing, and the overlapping sequences were combined. The sequence identity of the ORF and protein sequence were analyzed by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The sequence identities of the 3′-UTR and 3′-UTR variable region were analyzed by Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
2.6. Phylogenetic Analysis
Genome sequences of TMUVs were aligned, edited, and analyzed using Clustal W software [20]. The complete open reading frame sequences of TMUVs including sequences representing the most closely related strains to the TMUV strain TP1906 isolated in this study were obtained using BLAST and retrieved from GenBank for phylogenetic analysis (Table 2). The phylogenetic analysis was performed using MEGA version 7 (http://www.megasoftware.net/) [21]. To construct the phylogenetic trees, the maximum likelihood method using the Tamura–Nei model (Figures 3 and 5) and the maximum likelihood method using the JTT matrix-based model (Figure 4) were utilized. The reliability of the analysis was evaluated by a bootstrap test with 1000 replications. Sequences of Bagaza virus (GenBank numbers: KR108245 and MF380425), Japanese encephalitis virus (GenBank number: KF667310) and West Nile virus (GenBank number: M12294) were used as the outgroup.
3. Results
3.1. Identification of Novel TMUV Strains from Culex Mosquitoes in Taiwan
Mosquitoes were collected from nine localities in Taiwan from April to September 2019 (Figure 1). A total of 15,374 mosquitoes were collected and analyzed. Table 3 shows a summary of the mosquito species, numbers and RT-PCR results. Nineteen mosquito species from nine genera of the Culicidae family were identified. The most commonly collected species was Cx. tritaeniorhychus (70.90%, n = 10,900), followed by Cx. annulus (11.87%, n = 1825) and Mansonia uniformis (5.45%, n = 838). A flavivirus-specific primer set targeting the NS5 gene (amplicon is approximately 200 bp) was initially used for screening the mosquito pools by real-time RT-PCR. A total of 429 mosquito pools were tested, and 55 pools were positive; RT-PCR products were then sequenced and analyzed using the NCBI BLAST platform. Of the 55 positive pools, 53 pools contained genomic Japanese encephalitis virus (JEV) sequences, while a Cx. annulus mosquito pool (TP1906) collected from a wetland habitat for waterbirds in the Beitou District, Taipei City, in northern Taiwan, and a Cx. tritaeniorhynchus mosquito pool (TC1906) collected from a pig farm near rice paddy fields in Wufeng Township, Taichung City in central Taiwan, contained TMUV-like sequences. The partial NS5 gene sequences of RT-PCR products detected in the TP1906 and TC1906 mosquito pools were 100% identical and are closely related to STWV (GenBank number: JX477686) and TMUV-MM1775 (GenBank number: MH414569) sequences with 92.9% and 92.31% nucleotide similarities, respectively.
To further study whether the novel flaviviruses detected in Cx. annulus and Cx. tritaeniorhychus mosquito pools were identical, we amplified a partial NS1 gene sequence (964 bp) by using a TMUV NS1-specific primer set (TMUV-NS1_F and TMUV-NS1_R, in Table 1) [19]. The amplified NS1 gene sequences from these two mosquito pools were 99.79% identical with two nucleotide differences. The sequence obtained from the Cx. tritaeniorhynchus mosquito pool (TC1906) had a serine at amino acid position 131 of the NS1 protein (GenBank number: MN958524), while the Cx. annulus mosquito pool (TP1906) had an asparagine at position 131 (GenBank number: MN747003). The results indicated that the viruses from these two mosquito pools were very similar.
3.2. Isolation of a Novel TMUV from the Cx. Annulus Mosquito Pool
The C6/36 and Vero cell lines were used for propagation of the novel TMUV from the TP1906 and TC1906 mosquito pools. The novel TMUV strain TP1906 was successfully isolated from the Cx. annulus mosquito pool (TP1906). Virus isolation from the Cx. tritaeniorhynchus mosquito pool (TC1906) was unsuccessful.
3.3. Different Cell Lines Were Permissive for TMUV-TP1906 Infection
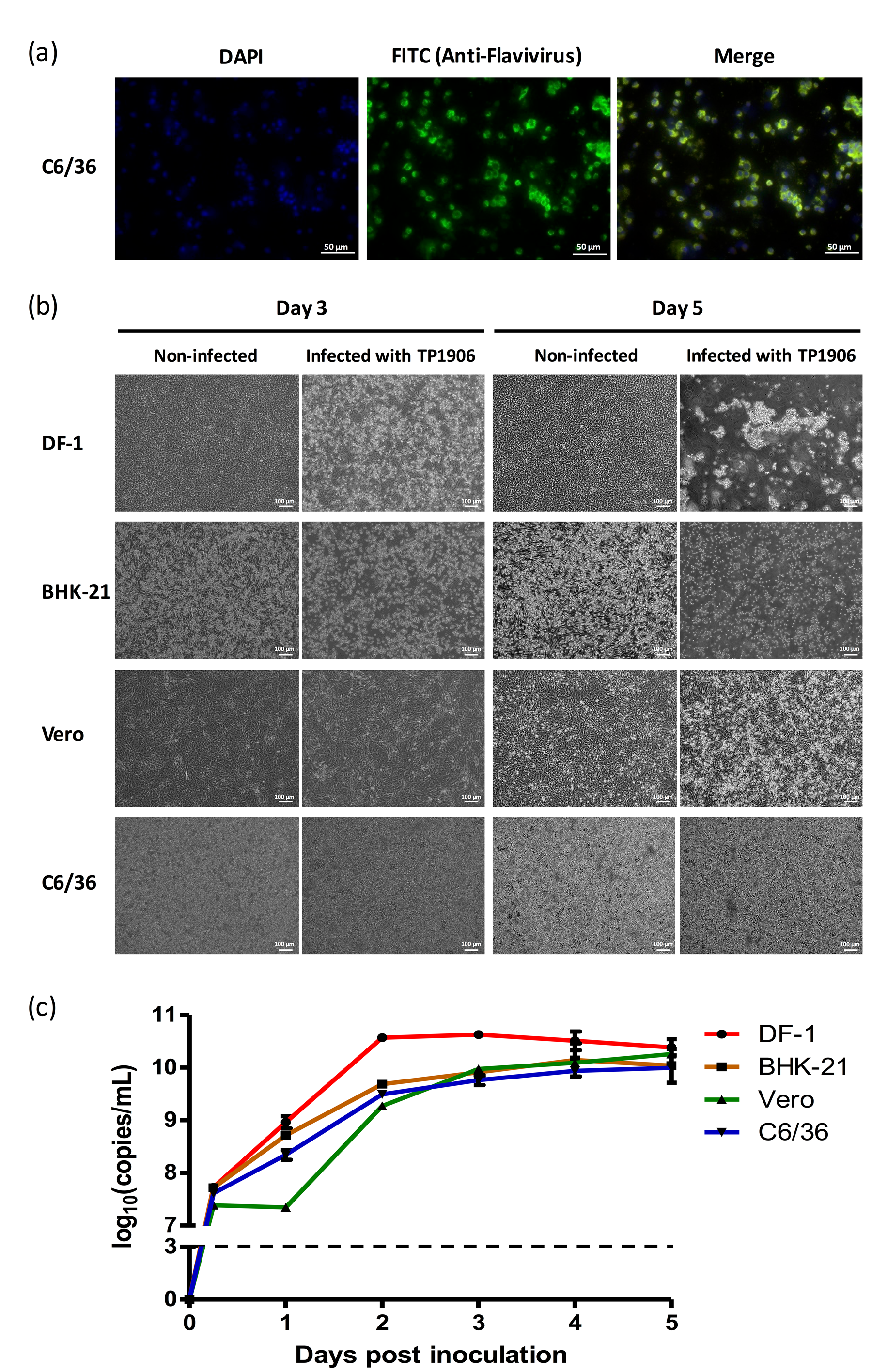
Immunofluorescence assay using anti-flavivirus antibody indicated that the TMUV-TP1906 was capable of infecting C6/36 cells (Figure 2a). To determine whether the TMUV-TP1906 replication can take place in different cell lines derived from mosquito (C6/36), monkey (Vero), hamster (BHK-21) and chicken (DF-1), we inoculated the TMUV-TP1906 to these cell lines and observed the cytopathic effects and detected viral RNA by using real-time RT-PCR. Although lacking a noticeable cytopathic effect (CPE) in C6/36 and Vero cell cultures, we observed apparent CPE in DF-1 and BHK-21 cell lines 72 h postinfection (Figure 2b). Notably, all the infected DF-1 and BHK-21 cells were detached five days postinfection. The TMUV-TP1906 was capable of replication in DF-1 cells and reached its highest titer at 48 h postinfection (Figure 2c). In contrast, the virus replication was relatively slowly in Vero cells compared to the BHK-21 and C6/36 cells.
3.4. Characteristics of the Genome Sequence of TMUV Strain TP1906
The 10990-nucleotide-long whole genome sequence of TMUV-TP1906 (GenBank number: MN747003) was determined to contain a 5′- UTR (94 nt), capsid(C; 360 nt), pre-membrane protein (prM; 501 nt), envelope (E; 1503 nt), nonstructural protein 1 (NS1; 1056 nt), NS2A (681 nt), NS2B (393 nt), NS3 (1857 nt), NS4A (378 nt), 2K (69 nt), NS4B (762 nt), NS5 (2718 nt); and 3′-UTR (618 nt). The sequence length of the ORF of TMUV-TP1906 was similar to that of other TMUVs, but the lengths of the 5′-UTR and 3′-UTR varied in different TMUVs. Table 4 shows comparative analyses of multiple alignments of different gene regions between TMUV-TP1906 and other TMUVs, including the STWV, TMUV-MM1775, DTMUV-SD14 and DTMUV strains. The results indicated that the ORF sequence of TMUV-TP1906 had 93.71%, 91.27% and 88% similarities to those of the STWV, TMUV-MM1775 and DTMUV-SD14 strains, respectively. The 5′-UTR (94 nt) sequence of TMUV-TP1906 had 94.68% and 92.55% similarities to those of the TMUV-MM1775 and DTMUV-SD14 strains, respectively. The 3′-UTR (618 nt) sequence was more conserved among TMUVs and had 95.15% and 92.72% similarity to the TMUV-MM1775 and DTMUV-SD14 strains, respectively.
3.5. Phylogenetic Analysis of TMUV-TP1906 and Other Flaviviruses Based on ORF Sequences
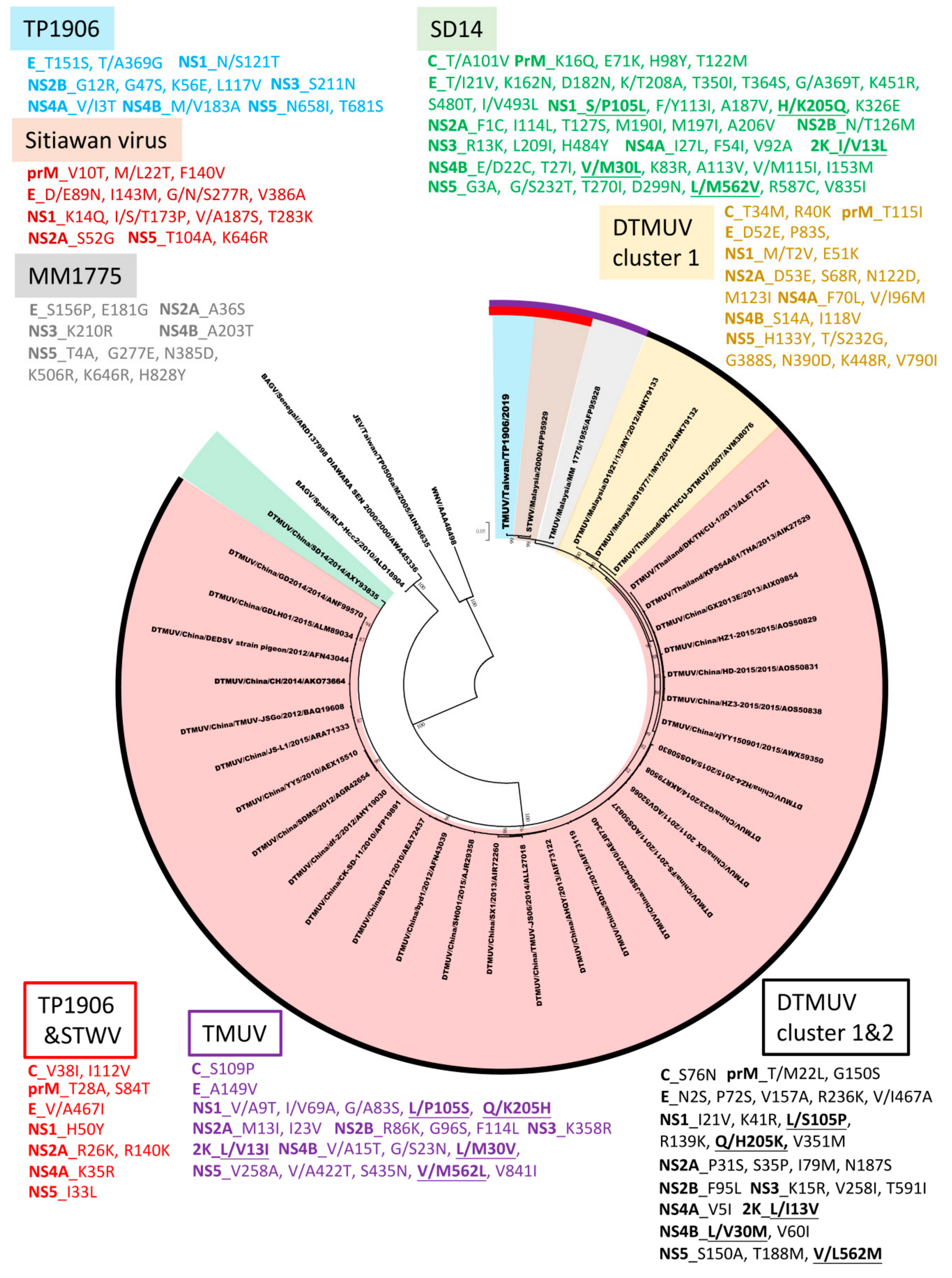
To understand the phylogenetic relationship between TMUV-TP1906 and other mosquito-borne flaviviruses, the complete ORF sequences of 40 representative flaviviruses, namely 36 TMUVs (listed in Table 2), Bagaza virus (GenBank numbers: KR108245 and MF380425), Japanese encephalitis virus (GenBank number: KF667310) and West Nile virus (GenBank number: M12294), were analyzed. Phylogenetic analysis indicated that TMUVs could be divided into two lineages (lineages TMUV and DTMUV). The TMUV lineage contained TMUV-MM1775, STWV, and TMUV-TP1906, and TMUV-TP1906 was most closely related to STWV. The DTMUV lineage could be further grouped into three clusters (clusters 1 to 3) (Figure 3). Cluster 1 contained DTMUV strains from Malaysia and Thailand. Clusters 2.1 and 2.2 contained many avian DTMUV strains found in Thailand and China. DTMUV-SD14 isolated from mallards (Anas platyrhynchos) in China was classified into cluster 3.
3.6. Amino Acid Sequence Comparison between TMUV-TP1906 and Other TMUVs
Genomic sequences of TMUV-TP1906 and other TMUV strains contain a single ORF that encodes a polypeptide of 3425 amino acids, except the ORFs of two DTMUV strains (D1977 and D1921) that encode 3424 amino acids with a deletion in the NS5 protein at amino acid position 86. The ORFs of TMUV-TP1906 and other TMUVs shared 86.52%–93.71% similarity at the nucleotide level and 95.71%–98.92% similarity when the amino acid sequences of the polyproteins were compared (Table 4). The amino acid sequences of the prM, E, and NS1 proteins of TMUV-TP1906 showed the highest similarity with TMUV-MM1775, while sequences of the C, NS2A, NS3, NS4A, NS4B, and NS5 proteins of TMUV-TP1906 showed the highest similarity with STWV. Figure 4 shows the phylogenetic relationship based on the amino acid sequences of polyproteins of TMUV-TP1906 and TMUVs. The results indicated that TMUV-TP1906 is most closely related to STWV.
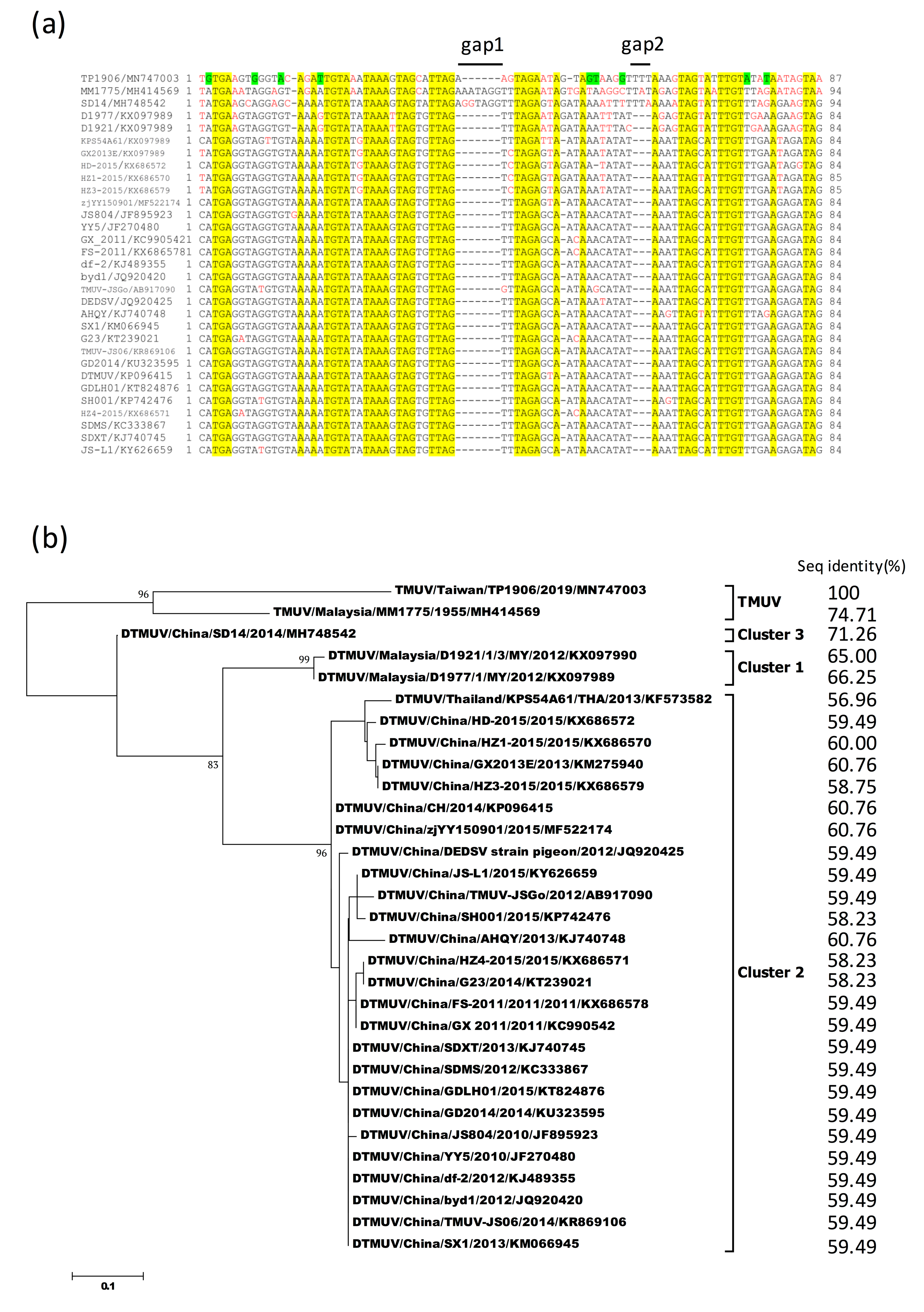
3.7. Sequence Alignment and Phylogenetic Analysis of 3′-UTR Variable Region of TMUV-TP1906 and Other TMUVs
Comparison of 5′-UTR and 3′-UTR sequences between TMUV-TP1906 and other TMUVs showed 90.43%–94.68% and 89.28%–95.15% sequence similarities, respectively (Table 4). Analysis of 3′-UTR variable region (VR) sequences (lengths varied from 84 to 94 nucleotides) between TMUV-TP1906 and other TMUVs showed that TMUV-TP1906 contained 1 gap (gap 1) and DTMUVs contained 2 gaps (gaps 1 and 2), while no gaps were found in the VR sequences of TMUV-MM1775 and DTMUV-SD14 (Figure 5a). The novel TMUV-TP1906 had a 3 nucleotide insertion in gap 2 and a nucleotide insertion in gap 1. In addition, there were nine unique nucleotide substitutions in the VR of TMUV-TP1906 compared with those of the other TMUVs. The 3′-UTR VR sequences were more consistent among cluster 2 of the DTMUV lineage, but more variable in the TMUV lineage and clusters 1 and 3 of the DTMUV lineage (Figure 5b). Comparisons of the 3′-UTR VR sequence of TMUV-TP1906 with those of TMUV-MM1775, DTMUV-SD14 (cluster 3), DTMUV-D1921 (cluster 1), and DTMUV-D1977 (cluster 1) showed 74.71%, 71.26%, 65.00%, and 66.25% sequence similarities, respectively, and 56.96%–60.76% sequence similarity with cluster 2 of DTMUVs. Figure 5b shows the phylogenetic tree based on the 3′-UTR VR sequences of TMUVs, and the overall topology was similar to the phylogenetic tree based on analysis of ORF sequences (Figure 3).
4. Discussion
In this study, we identified two novel TMUV strains, TMUV-TP1906 and TMUV-TC1906, from Cx. annulus and Cx. tritaeniorhynchus mosquitoes collected from a wetland in northern Taiwan and a pig farm in central Taiwan, respectively. TMUV-TP1906 was isolated from the Cx. annulus mosquito pool and grew well in Vero and C6/36 cells without significant cytopathic effects. Through virus isolation and comprehensive genomic and protein sequence analysis, we found that this novel flavivirus is a TMUV-related virus and is most closely related to STWV. STWV, DTMUV, and Bagaza virus, which belong to Ntaya serocomplex virus; and JEV and West Nile virus, which belong to JEV serocomplex virus, have been shown to be primarily Culex mosquito-associated viruses that cause severe diseases in avian species [7]. Whether TMUV-TP1906 is a pathogen in birds or other animals needs further study.
Phylogenetic analysis based on the complete ORF of TMUVs showed that TMUVs can be divided into two lineages, TMUV and DTMUV. Analysis of amino acid sequences showed that there are 22 amino acid substitutions unique to the TMUV lineage and 29 amino acid substitutions unique to cluster 1 and cluster 2 of the DTMUV lineage (Figure 4). The DTMUV-SD14 strain which belongs to cluster 3 of the DTMUV lineage, could be considered as a transitional group between the TMUV lineage and clusters 1 and 2 of the DTMUV lineage. The DTMUV-SD14 strain contains 48 amino acid mutations, which are different from other TMUVs. In addition, there are five informative amino acid substitutions (NS1-105, NS1-205, 2K-13, NS4B-30 and NS5-562) for the differentiation of the TMUV lineage, DTMUV-SD14 (cluster 3 of the DTMUV lineage) and clusters 1 and 2 of the DTMUV lineage. The TMUV lineage contains three virus strains, the TMUV-MM1775 prototype strain, STWV, and TMUV-TP1906. TMUV-MM1775 has not been documented as a pathogen in birds or other animals; however, STWV, which is most closely related to TMUV-TP1906, causes encephalitis and retards the growth of chicks. Analysis of amino acid sequences showed that TMUV-TP1906 contains 12 unique amino acid substitutions compared with other TMUVs. In addition, comparison of protein sequences of TMUV-TP1906 and STWV with other TMUVs showed 10 amino acid substitutions. These informative amino acid positions may be useful for the classification of TMUVs and may play crucial roles in the evolution and protein function of TMUVs.
The Asn glycosylation site at residue 154 of the E protein (154-Asn) of TMUVs is critical for virus tissue tropism and transmissibility in poultry [24]. TMUV-MM1775 contains 156-Pro, which can disrupt the N-linked glycosylation of 154-Asn of the E protein, and thus may affect viral virulence and transmissibility in avian species [24]. Except for TMUV-MM1775, all the other TMUVs, including TMUV-TP1906, contain 156-Ser which would not disrupt 154-Asn glycosylation and thus may not affect the glycosylation-associated infectivity of TMUVs in avian species. A recent study showed that the Thr to Lys mutation at residue 367 of the E protein plays a predominant role in viral cell-adaptation and virulence attenuation in ducks [25]. TMUV-TP1906 contains 367-Thr of the E protein and thus may retain the virulence in the host. In addition to E protein, NS1 protein contains three N-linked glycosylation sites at residues 130, 175, and 207. A recent study showed that some DTMUV strains had a glycosylation motif mutation at residues 175–178 from NTTD to NITD but the glycosylation site at 130 was conserved [19]. Interestingly, we found NS1 glycosylation motif at residues 175 and 207 were conserved in TMUV-TP1906 and TMUV-TC1906, however, TMUV-TC1906 had a glycosylation motif mutation at residues 130–133 from NNTF to NSTF. Whether these mutations affect protein functions or virulence needs further study.
The 5′ and 3′ UTRs of flaviviruses are important for virus replication and transmission [26]. The 5′ end of the 3′UTR of flavivirus is known to have a VR with different nucleotide lengths [26], and this VR sequence may be associated with the production of subgenomic flavivirus RNA and immune modulation, hence contributing to virus adaptation in vector and non-vector hosts [27,28]. The 3′UTR VRs of TMUVs consist of 84 to 94 nucleotides immediately downstream of the ORF. Phylogenetic analysis of 3′UTR VR and ORF sequences showed similar topological features, indicating that 3′UTR VRs may serve as a potential marker for TMUV evolution. Sequence analysis also showed that the 3′UTR VR of TMUV-TP1906 is very unique among TMUVs (Figure 5). More research is needed to understand the genetic diversity and function of the 3′UTR VR of TMUVs.
Prominent CPE was observed in BHK-21, C6/36, Vero and DF-1 cell lines after infection with DTMUV [5,10,18,29]. STWV infection induced CPE in the BK3 cells (chicken bursal lymphoma cell line) but not in CPK (porcine kidney cell line), MARC-145 (monkey kidney cell line) and Vero cells [2]. In our study, we found that the TMUV-TP1906, which is closely related to STWV, grew in C6/36 and Vero cell without inducing significant CPE, however, the virus caused drastic CPE in DF-1 and BHK-21 cell lines (Figure 2).
TMUVs are emerging pathogenic flaviviruses causing severe avian diseases in Malaysia, Thailand, and China. Taiwan is located south of East Asia and is close to the epicenter of DTMUV outbreaks in China. In this study, we first identified a TMUV in Taiwan. Interestingly, TMUV-TP1906 is most closely related to STWV and TMUV-MM1775 found in Malaysia and is different from the DTMUVs found in China (Figure 3 and Figure 4 Figure 3; Figure 4). Previous study has divided TMUVs into the Chinese mainland TMUV lineage and Southeast Asian TMUV lineage [8]. According to the phylogeny, TMUV-MM1775, STWV, TMUV-TP1906, and two TMUV strains identified from Cx. tritaeniorhynchus in Yunnan Province of China were grouped as the Southeast Asian TMUV lineage. The phylogeographical analysis suggested that the TMUVs might spread from Southeast Asian countries such as Malaysia and Thailand to the Yunnan Province of China (southern China) and further spread to areas in northern China such as Shandong Province. However, the mechanism of long-distance spread of TMUVs is still unknown. Liu et al. suggested that the spread of TMUVs might occur via migratory birds and ornithophilic Culex spp. mosquitoes, since TMUVs have been identified in wild birds such as pigeons and mallards (Anas platyrhynchos) [7]. The East Asian–Australasian flyway is one of the world’s great flyways for migratory birds, and the flyway passes through many countries including Malaysia, Thailand, China, and Taiwan; thus, it is possible that the spread of TMUVs might be through this flyway. In addition, it has been suggested that the spread of avian influenza virus and JEV might also occur through this flyway [30,31].
In this study, we reported the first isolation of a novel TMUV strain from Culex mosquitoes in Taiwan. In addition, it is the first time that the TMUV strain was isolated from Cx. annulus mosquitoes. We performed comprehensive genomic and amino acid analyses between TMUV-TP1906 and other TMUVs, and our results may help to increase understanding of the diversity and evolution of TMUVs. Further study is warranted to investigate the virulence and epidemiology of TMUV-TP1906 in Taiwan.
Author Contributions
Conception and design of experiments, S.-H.P., C.-L.S., and P.-Y.S.; methodology and performing of experiments, S.-H.P., C.-L.S., M.-C.C., and H.-C.H.; data analysis, S.-H.P., C.-L.S., M.-C.C., H.-C.H., S.-L.Y., and P.-Y.S.; Writing of paper, S.-H.P. and P.-Y.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MOHW108-CDC-C-315-122401 and MOHW109-CDC-C-315-113107 from Centers for Disease Control, Ministry of Health and Welfare, Taiwan, Republic of China.
Acknowledgments
The authors would like to thank the members of Vector-Borne Viral and Rickettsia Disease Lab in Taiwan Centers for Disease Control for collecting the mosquitoes and Liang-Chen Lu for classification of the mosquito species.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results
References
- Benzarti, E.; Linden, A.; Desmecht, D.; Garigliany, M. Mosquito-borne epornitic flaviviruses: An update and review. J. Gen. Virol. 2019, 100, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kono, Y.; Tsukamoto, K.; Abd Hamid, M.; Darus, A.; Lian, T.C.; Sam, L.S.; Yok, C.N.; Di, K.B.; Lim, K.T.; Yamaguchi, S.; et al. Encephalitis and retarded growth of chicks caused by Sitiawan virus, a new isolate belonging to the genus Flavivirus. Am. J. Trop. Med. Hyg. 2000, 63, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Li, S.; Hu, X.; Yu, X.; Wang, Y.; Liu, P.; Lu, X.; Zhang, G.; Hu, X.; Liu, D.; et al. Duck egg-drop syndrome caused by BYD virus, a new Tembusu-related flavivirus. PLoS ONE 2011, 24, e18106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Zhang, C.; Liu, Y.; Liu, Y.; Ye, W.; Han, J.; Ma, G.; Zhang, D.; Xu, F.; Gao, X.; et al. Tembusu Virus in Ducks, China. Emerg. Infect. Dis. 2011, 17, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Homonnay, Z.G.; Kovács, E.W.; Bányai, K.; Albert, M.; Fehér, E.; Mató, T.; Tatár-Kis, T.; Palya, V. Tembusu-like flavivirus (Perak virus) as the cause of neurological disease outbreaks in young Pekin ducks. Avian Pathol. 2014, 43, 552–560. [Google Scholar] [CrossRef]
- Thontiravong, A.; Ninvilai, P.; Tunterak, W.; Nonthabenjawan, N.; Chaiyavong, S.; Angkabkingkaew, K.; Mungkundar, C.; Phuengpho, W.; Oraveerakul, K.; Amonsin, A. Tembusu-Related Flavivirus in Ducks, Thailand. Emerg. Infect. Dis. 2015, 21, 2164–2167. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Lu, H.; Li, S.; Moureau, G.; Deng, Y.Q.; Wang, Y.; Zhang, L.; Jiang, T.; de Lamballerie, X.; Qin, C.F.; et al. Genomic and antigenic characterization of the newly emerging Chinese duck egg-drop syndrome flavivirus: Genomic comparison with Tembusu and Sitiawan viruses. J. Gen. Virol. 2012, 93 Pt 10, 2158–2170. [Google Scholar] [CrossRef] [Green Version]
- Lei, W.; Guo, X.; Fu, S.; Feng, Y.; Tao, X.; Gao, X.; Song, J.; Yang, Z.; Zhou, H.; Liang, G. The genetic characteristics and evolution of Tembusu virus. Vet. Microbiol. 2017, 201, 32–41. [Google Scholar] [CrossRef]
- Pandey, B.D.; Karabatsos, N.; Cropp, B.; Tagaki, M.; Tsuda, Y.; Ichinose, A.; Igarashi, A. Identification of a flavivirus isolated from mosquitos in Chiang Mai Thailand. Southeast Asian J. Trop. Med. Public Health 1999, 30, 161–165. [Google Scholar]
- Tang, Y.; Diao, Y.; Chen, H.; Ou, Q.; Liu, X.; Gao, X.; Yu, C.; Wang, L. Isolation and genetic characterization of a tembusu virus strain isolated from mosquitoes in Shandong, China. Transbound. Emerg. Dis. 2015, 62, 209–216. [Google Scholar] [CrossRef]
- O’Guinn, M.L.; Turell, M.J.; Kengluecha, A.; Jaichapor, B.; Kankaew, P.; Miller, R.S.; Endy, T.P.; Jones, J.W.; Coleman, R.E.; Lee, J.S. Field detection of Tembusu virus in western Thailand by RT-PCR and vector competence determination of select Culex mosquitoes for transmission of the virus. Am. J. Trop. Med. Hyg. 2013, 89, 1023–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Shi, Y.; Liu, Q.; Wang, Y.; Li, G.; Teng, Q.; Zhang, Y.; Liu, S.; Li, Z. Airborne transmission of a novel Tembusu virus in ducks. J. Clin. Microbiol. 2015, 53, 2734–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, P.; Zhao, Y.; Zhang, X.; Xu, D.; Dai, X.; Teng, Q.; Yan, L.; Zhou, J.; Ji, X.; Zhang, S.; et al. An infectious disease of ducks caused by a newly emerged Tembusu virus strain in mainland China. Virology 2011, 417, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birding Taiwan. Available online: https://taiwantoday.tw/news.php?unit=14&post=23956&unitname=Environment-Taiwan-Review&postname=Birding-Taiwan (accessed on 1 August 2013).
- Su, C.L.; Yang, C.F.; Teng, H.J.; Lu, L.C.; Lin, C.; Tsai, K.H.; Chen, Y.Y.; Chen, L.Y.; Chang, S.F.; Shu, P.Y. Molecular epidemiology of Japanese encephalitis virus in mosquitoes in Taiwan during 2005–2012. PLoS Negl. Trop. Dis. 2014, 8, e3122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.H.; Lin, T.H.; Teng, H.J.; Su, C.L.; Tsai, K.H.; Lu, L.C.; Lin, C.; Yang, C.F.; Chang, S.F.; Liao, T.L.; et al. Molecular epidemiology of Japanese encephalitis virus, Taiwan. Emerg. Infect. Dis. 2010, 16, 876–878. [Google Scholar] [CrossRef]
- Shu, P.Y.; Chang, S.F.; Kuo, Y.C.; Yueh, Y.Y.; Chien, L.J.; Sue, C.L.; Lin, T.H.; Huang, J.H. Development of group- and serotype-specific one-step SYBR green I-based real-time reverse transcription-PCR assay for dengue virus. J. Clin. Microbiol. 2003, 41, 2408–2416. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Han, K.; Zhao, D.; Liu, Y.; Zhang, J.; Niu, H.; Zhang, K.; Zhu, J.; Wu, D.; Gao, L.; et al. Identification and molecular characterization of a novel flavivirus isolated from geese in China. Res. Vet. Sci. 2013, 94, 774–780. [Google Scholar] [CrossRef]
- Yu, G.; Lin, Y.; Tang, Y.; Diao, Y. Evolution of Tembusu virus in ducks, chikens, geeses, sparrows, and mosquitoes in northern China. Viruses 2018, 10, 485. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Shi, Y.; Wang, H.; Li, G.; Li, X.; Wang, B.; Su, X.; Wang, J.; Teng, Q.; Yang, J.; et al. A single mutation at position 156 in the envelope protein of Tembusu virus is responsible for virus tissue tropism and transmissibility in ducks. J. Virol. 2018, 16, e00427-18. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhang, L.; Cao, Y.; Wang, J.; Yu, Z.; Sun, X.; Liu, F.; Li, Z.; Liu, P.; Su, J. Basic amino acid substitution at residue 367 of Tembusu virus plays critical role in pathogenesis. J. Virol. 2020, 31, e02011-19. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.C.; Soto-Acosta, R.; Bradrick, S.S.; Garcia-Blanco, M.A.; Ooi, E.E. The 5′ and 3′ untranslated regions of the flaviviral genome. Viruses 2017, 6, 137. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, D.E.; De Lella Ezcurra, A.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3′-UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Villordo, S.M.; Filomatori, C.V.; Sanchez-Vargas, I.; Blair, C.D.; Gamarnik, A.V. Dengue virus RNA structure specialization facilities host adaption. PLoS Pathog. 2015, 30, e1004604. [Google Scholar] [CrossRef]
- Wang, H.J.; Li, X.F.; Liu, L.; Xu, Y.P.; Ye, Q.; Deng, Y.Q.; Huang, X.Y.; Zhao, H.; Qin, E.D.; Shi, P.Y.; et al. The emerging duck flavivirus is not pathogenic for primates and is highly sensitive to mammalian interferon antiviral signaling. J. Virol. 2016, 90, 6538–6548. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.C.; Lee, M.S.; Ho, Y.H.; Chyi, W.L.; Wang, C.H. Avian influenza monitoring in migrating in Taiwan during 1998–2007. Avian Dis. 2010, 54, 109–114. [Google Scholar] [CrossRef]
- Gao, X.; Liu, H.; Wang, H.; Fu, S.; Guo, Z.; Liang, G. Southernmost Asia is the source of Japanese encephalitis virus (genotype 1) diversity from which the viruses disperse and evolve throughout Asia. PLoS Negl. Trop. Dis. 2013, 19, e2459. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Map showing mosquito collection sites in Taiwan. Mosquitoes collected from wetland, parks and pig farms are indicated with blue, green and red colors, respectively. Black arrows indicate collection sites of mosquitoes infected with novel TMUV strains TP1906 and TC1906.
Figure 1.
Map showing mosquito collection sites in Taiwan. Mosquitoes collected from wetland, parks and pig farms are indicated with blue, green and red colors, respectively. Black arrows indicate collection sites of mosquitoes infected with novel TMUV strains TP1906 and TC1906.

Figure 2.
Different cell lines were permissive to TMUV-TP1906 infection. (a) The TMUV-TP1906 infection was detected by IFA in C6/36 cells. (b) The morphology of TMUV-TP1906 infected cells at 3 days and 5 days postinfection. (c) Growth curves of TMUV-TP1906 in different cell lines. All the experiments were performed in triplicate. Dashed line indicates the limit of detection of real time RT-PCR by using the flavivirus-specific primer set.
Figure 2.
Different cell lines were permissive to TMUV-TP1906 infection. (a) The TMUV-TP1906 infection was detected by IFA in C6/36 cells. (b) The morphology of TMUV-TP1906 infected cells at 3 days and 5 days postinfection. (c) Growth curves of TMUV-TP1906 in different cell lines. All the experiments were performed in triplicate. Dashed line indicates the limit of detection of real time RT-PCR by using the flavivirus-specific primer set.

Figure 3.
Phylogeny of TMUV-TP1906, Tembusu virus strains (TMUVs) and duck Tembusu virus strains (DTMUVs) based on the ORF (10,278 nt). The evolutionary history was inferred by using the maximum likelihood method based on the Tamura–Nei model [22]. The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 40 nucleotide sequences. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA7 [21]. The reliability of the analysis was calculated using 1000 bootstrap replication. Bootstrap support values >75 are shown. The scale bar indicates nucleotide substitutions per site.
Figure 3.
Phylogeny of TMUV-TP1906, Tembusu virus strains (TMUVs) and duck Tembusu virus strains (DTMUVs) based on the ORF (10,278 nt). The evolutionary history was inferred by using the maximum likelihood method based on the Tamura–Nei model [22]. The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 40 nucleotide sequences. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA7 [21]. The reliability of the analysis was calculated using 1000 bootstrap replication. Bootstrap support values >75 are shown. The scale bar indicates nucleotide substitutions per site.

Figure 4.
Phylogeny of TMUV-TP1906, Tembusu virus strains (TMUVs), and duck Tembusu virus strains (DTMUVs) based on the polyprotein amino acid sequences (3425 aa). The evolutionary history was inferred by using the maximum likelihood method based on the JTT matrix-based model [23]. The tree with the highest log likelihood is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 40 amino acid sequences. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA7 [21]. The reliability of the analysis was calculated using 1000 bootstrap replication. Bootstrap support values >75 are shown. The scale bar indicates amino acid substitutions per site. The unique amino acid residues in TP1906, Sitiawan virus, MM1775, SD14, and DTMUV cluster 1 compared to other TMUV strains were colored in blue, brown, gray, green, and yellow, respectively. The unique amino acid residues in DTMUV cluster 1 and 2, TMUV lineage, and TP1906 and STWV compared to other TMUV strains were colored in black, purple, and red, respectively.
Figure 4.
Phylogeny of TMUV-TP1906, Tembusu virus strains (TMUVs), and duck Tembusu virus strains (DTMUVs) based on the polyprotein amino acid sequences (3425 aa). The evolutionary history was inferred by using the maximum likelihood method based on the JTT matrix-based model [23]. The tree with the highest log likelihood is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 40 amino acid sequences. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA7 [21]. The reliability of the analysis was calculated using 1000 bootstrap replication. Bootstrap support values >75 are shown. The scale bar indicates amino acid substitutions per site. The unique amino acid residues in TP1906, Sitiawan virus, MM1775, SD14, and DTMUV cluster 1 compared to other TMUV strains were colored in blue, brown, gray, green, and yellow, respectively. The unique amino acid residues in DTMUV cluster 1 and 2, TMUV lineage, and TP1906 and STWV compared to other TMUV strains were colored in black, purple, and red, respectively.

Figure 5.
Sequence alignment and phylogenetic tree of 3′UTR variable regions of TMUV-TP1906, Tembusu virus strains (TMUVs), and duck Tembusu virus strains (DTMUVs). (a) 3′UTR sequence alignment of TMUV-TP1906, TMUVs, and DTMUVs. The conserved nucleotides are highlighted in yellow and the unique nucleotides of TMUV-TP1906 are highlighted in green. The nucleotides different from the consensus sequence are colored in red; (b) phylogeny of TMUV-TP1906, TMUVs and DTMUVs based on 3′UTR variable region. The evolutionary history was inferred by using the maximum likelihood method based on the Tamura–Nei model. The reliability of the analysis was calculated using 1000 bootstrap replication. Bootstrap support values >75 are shown. The scale bar indicates amino acid substitutions per site. Sitiawan virus and four DTMUV strains including DK/TH/CU-1, BYD-1, CK-SD-11, and DK/TH/CU-DTMUV which do not deposit the 3′-UTR sequences in GenBank were not analyzed.
Figure 5.
Sequence alignment and phylogenetic tree of 3′UTR variable regions of TMUV-TP1906, Tembusu virus strains (TMUVs), and duck Tembusu virus strains (DTMUVs). (a) 3′UTR sequence alignment of TMUV-TP1906, TMUVs, and DTMUVs. The conserved nucleotides are highlighted in yellow and the unique nucleotides of TMUV-TP1906 are highlighted in green. The nucleotides different from the consensus sequence are colored in red; (b) phylogeny of TMUV-TP1906, TMUVs and DTMUVs based on 3′UTR variable region. The evolutionary history was inferred by using the maximum likelihood method based on the Tamura–Nei model. The reliability of the analysis was calculated using 1000 bootstrap replication. Bootstrap support values >75 are shown. The scale bar indicates amino acid substitutions per site. Sitiawan virus and four DTMUV strains including DK/TH/CU-1, BYD-1, CK-SD-11, and DK/TH/CU-DTMUV which do not deposit the 3′-UTR sequences in GenBank were not analyzed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Primers used in RT-PCR and DNA sequencing for TMUV-TP1906.
| Name | Sequence (5′ to 3′) | Amplicon (bp) | Annealing Temp (°C) | References |
|---|---|---|---|---|
| P1f | AGAAGTTCRYCTGTGTGA | 638 | 50 | [6,18] |
| DF_R638 | CAGCAGTCTATGTCTTCAGG | |||
| DF_F441 | CGATAGTTGCTGGGCTGAAGC | 675 | 50 | [6] |
| DF_R1115 | GCAGTAAGATCTCACAACCGC | |||
| DF_F954 | GCTTCAGCTGTCTGGGGATGC | 696 | 50 | |
| DF_R1650 | CAATGACTCTTTGTTTTGCCACG | |||
| DF_F2353 | GGCACTGCTATTGTGGATGGG | 987 | 50 | |
| DF_R3339 | GGTGGGGTGGTGCAAGACC | |||
| DF_F3302 | GGAACAACTGTCACAGTAACG | 1393 | 50 | |
| DF_R4694 | GCATGACTCCCACTCCAGCC | |||
| DF_F4406 | GCATCACAGAGATTTGATGTGG | 757 | 54 | |
| DF_R5162 | CCTGAACCTGGATGTAGGTCC | |||
| DF_F4874 | GCAAGTCATCGTCGTGCAACC | 709 | 50 | |
| DF_R5582 | GCTCTTCAATGTCTGTTATTGGC | |||
| DF_F5399 | GCTCACACCTCAGCGAGTGC | 851 | 50 | |
| DF_R6249 | GGTCATTGTAACTTATCCCAGC | |||
| DF_F6494 | CGCTCACAGAATGACAGAATCC | 855 | 50 | |
| DF_R7348 | GGAACATCTGTAGCCACTATGC | |||
| DF_F6807 | GAACCAGAGAGACAGAGATCGC | 1352 | 50 | |
| DF_R8158 | CCCTAGCTAGCCATTCCTCGG | |||
| DF_F7940 | GCAGGTTCAGGAAGTGAGAGG | 597 | 50 | |
| DF_R8536 | GGATTGTCTTGGTCATAATGCC | |||
| DF_F8383 | GGATGCACAAAACCAACCGC | 833 | 50 | |
| DF_R9215 | GGCCGAGATGTCACGCAGC | |||
| DF_F9274 | GGGACACTAGAATAACCAAGGC | 1212 | 50 | |
| DF_R10485 | CCAACATCCGGTGGCAGGG | |||
| TMUV-E_F | TTCAGCTGTCTGGGGATGCA | 1503 | 50 | [19] |
| TMUV-E_R | GGCATTGACATTTACTGCCA | |||
| TMUV-NS1_F | GACACGGGGTGCTCAATCGACTT | 1056 | 50 | |
| TMUV-NS1_R | AGCCATGACCTTTGATTTGAT | |||
| TMUV-NS3_F | GGAGGAGTCATCTGGGATGTG | 1857 | 50 | |
| TMUV-NS3_R | TCTCTTTCCACTCGCAAAATC | |||
| TMUV-NS5_F | GAACTGGCAGAACTTTGGGGGAG | 2711 | 50 | |
| TMUV-NS5_R | TTACAAGACACCTTCACTCCAGC | |||
| 1_F | AAATGACTTCAGGACACCTC | 723 | 50 | This study * |
| 1_R | ACATACCTTGTCCACACTTC | |||
| 2_F | ATGTCATGGATCACTCAAGG | 400 | 50 | |
| 2_R | CAGTCAAGTCAATGCTGTTG | |||
| 3_F | AAAAGAAAGGAGGCATGCTA | 523 | 50 | |
| 3_R | GGAACATCCCATATGACTCC | |||
| 5_F | CAGTCGGAAGTGCATTAAAC | 683 | 54 | |
| 5_R | CAGCTGTAGTCAGCATGTAT | |||
| 7_F | TTAGAATCCTGTCAAAGCCC | 767 | 50 | |
| 7_R | CACCTTCACCACCTTATTCA | |||
| 8_F | CTGGAATCTCGTTGATAGGG | 572 | 50 | |
| 8_R | TAATGAGTTGAACGCACAGA | |||
| 3′UTR-2_F | CCAACCCTCAATAGGTTCAA | 612 | 50 | This study † |
| 3′UTR-2_R | GAGGGTCTCCTAGTCTATCC | |||
| 3′UTR-5_F | ATACATGGAAGACAAGACCC | 465 | 50 | |
| 3′UTR-5_R | GAGACGGTATTGAACGCTTA | |||
| 3′UTR-10_F | AGGAGCTAAGCGTTCAATAC | 427 | 50 | |
| 3′UTR-10_R | GACTCTGTGTTCTACCACC | |||
| GSP-5′ end | GGTCGCCTCACTGACCCCAACTAGC | 331 | 55 | For RACE |
| 3′UTR-10_F | AGGAGCTAAGCGTTCAATAC | 424 | 55 | |
| oligo d(T)-anchor | GACCACGCGTATCGATGTCGACT(16)V | 55 |
* Design based on Sitiawan virus cDNA sequence (JX477686); † design based on M1775 virus cDNA sequence (MH414569).
Table 2.
Tembusu virus strains used for phylogenetic analysis in this study.
| TMUV /DTMUV Strains | Host | Location | Year/ Month | GenBank Accession Number /ORF Sequence Identity (%)/ Length (Nucleotides) | GenBank Accession Number /Protein Sequence Identity (%)/ Length (Amino Acids) |
|---|---|---|---|---|---|
| TP1906 | Cx. annulus | Taipei/ Taiwan | 2019/ Jun | MN747003 100 (10,990) | 100 (3425) |
| TC1906 † | Cx. tritaeniorhynchus | Taichung/ Taiwan | 2019/ Jun | MN958524 NS1:99.79 (964) | NS1:99.69 (964) |
| Sitiawan virus | Broiler Chicks | Perak state/ Malaysia | 2000 | JX477686 93.71 (10,278) | AFP95929 98.92 (3425) |
| MM1775 | Cx. tritaeniorhynchus | Kuala Lumpar/ Malaysia | 1955 | JX477685 91.27 (10,278) | AFP95928 98.57 (3425) |
| YY5 | Duck | Zhejiang Province/ China | 2010 | JF270480 87.05 (10,990) | AEX15510 96.50 (3425) |
| BYD-1 | Duck | China | 2010 | JF312912 87.15 (10,278) | AEA72437 96.55 (3425) |
| JS804 | Goose | Jiangsu Province/ China | 2010 | JF895923 87.12 (10,990) | AEJ87340 96.38 (3425) |
| CK-SD-11 | Chicken | China | 2010 | JQ627862 87.01 (10,278) | AFP19891 96.41 (3425) |
| GX_2011 | Duck | Guangxi Province/ China | 2011 | KC990542 87.05 (10,990) | AGV52066 96.47 (3425) |
| FS-2011 | Duck | China | 2011 | KX686578 87.00 (10,991) | AOS50837 96.44 (3425) |
| df-2 | Duck | China | 2012 | KJ489355 87.16 (10,990) | AHY19030 96.53 (3425) |
| byd1 | Duck | Hebei Province/ China | 2012 | JQ920420 87.15 (10,990) | AFN43039 96.55 (3425) |
| JSGo | Goose | China | 2012 | AB917090 87.21 (10,959) | BAQ19608 96.53 (3425) |
| DEDSV strain pigeon | Pigeon | Beijing Autonomous City/ China | 2012 | JQ920425 87.09 (10,990) | AFN43044 96.61 (3425) |
| AHQY | Layer Duck | China | 2013 | KJ740748 86.65 (10,990) | AIF73122 96.35 (3425) |
| SX1 | Chicken | China | 2013 | KM066945 86.80 (10,990) | AIR72260 95.97 (3425) |
| G23 | Goose | China | 2014 | KT239021 86.92 (10,881) | AKR79508 96.61 (3425) |
| JS06 | Chicken | China | 2014 | KR869106 86.69 (10,990) | ALL27018 95.7 (3425) |
| GD2014 | Duck | China | 2014 | KU323595 87.06 (10,990) | ANF99570 96.50 (3425) |
| DTMUV/CH/2014 | Duck | China | 2014 | KP096415 87.05 (10,990) | AKO73664 96.44 (3425) |
| GDLH01 | Duck | China | 2015 | KT824876 87.07 (10,990) | ALM89034 96.47 (3425) |
| SH001 | Duck | Shanghai Province /China | 2015 | KP742476 87.10 (10,990) | AJR29358 96.50 (3425) |
| HZ4-2015 | Broiler Duck | China | 2015 | KX686571 86.63 (10,991) | AOS50830 95.94 (3425) |
| SD14 | Anas platyrhynchos | China | 2014 | MH748542 88.00 (11,001) | AXY93835 96.64 (3425) |
| DK/TH/CU-DTMUV | Duck | Thailand | 2007 | MF621927 87.21 (10,278) | AVM38076 96.41 (3425) |
| D1977/1/MY | Pekin Duck | Malaysia | 2012 | KX097989 87.09 (10,988) | ANK79132 96.20 (3424) |
| D1921/1/3/MY | Pekin Duck | Malaysia | 2012 | KX097990 87.10 (10,988) | ANK79133 96.18 (3424) |
| DK/TH/CU-1 | Duck | Thailand | 2013 | KR061333 86.91 (10,278) | ALE71321 96.55 (3425) |
| KPS54A61 | Duck | Thailand | 2013 | KF573582 86.84 (10,990) | AIK27529 96.35 (3425) |
| GX2013E | Duck | China | 2013 | KM275940 86.97 (10,990) | AIX09854 96.23 (3425) |
| HD-2015 | Layer Duck | China | 2015 | KX686572 86.75 (10,991) | AOS50831 96.15 (3425) |
| HZ1-2015 | Layer Duck | China | 2015 | KX686570 86.52 (10,990) | AOS50829 96.12 (3425) |
| HZ3-2015 | Duck | China | 2015 | KX686579 86.94 (10,992) | AOS50838 96.18 (3425) |
| zjYY150901 | Duck | China | 2015 | MF522174 86.82 (10,990) | AWX59350 96.29 (3425) |
| SDMS | Culex Mosquito | Shangdong Province/China | 2012 | KC333867 87.04 (10,990) | AGR42654 96.47 (3425) |
| SDXT | Layer Duck | China | 2013 | KJ740745 87.08 (10,990) | AIF73119 96.58 (3425) |
| JS-L1 | Duck | China | 2015 | KY626659 87.14 (10,890) | ARA71333 96.53 (3425) |
† The sequence identity of TC1906 partial NS5 (156 nt) and NS1 (964 nt) was 100% and 99.79% with TP1906, respectively.
Table 3.
Summary of the mosquito species, number of mosquitoes, and pools tested and positive Tembusu virus pools.
Table 3.
Summary of the mosquito species, number of mosquitoes, and pools tested and positive Tembusu virus pools.
| Species | Taipei | Taichung | Tainan | Yilan | Hualien | Num Individuals | Num Pools | Num TMUV Pos Pools | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Park | Wetland | Park | Pig Farm | Park | Pig Farm | Park | Pig Farm | Pig Farm | ||||
| Aedes aegypti | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 7 | 3 | 0 |
| Aedes albopictus | 86 | 110 | 4 | 0 | 33 | 0 | 43 | 0 | 33 | 309 | 23 | 0 |
| Aedes malikuli | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 3 | 0 |
| Aedes penghuensis | 0 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 4 | 0 |
| Aedes vexans | 0 | 75 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 75 | 5 | 0 |
| Anopheles sinensis | 0 | 0 | 0 | 106 | 0 | 3 | 0 | 12 | 35 | 156 | 8 | 0 |
| Anopheles tessellatus | 0 | 179 | 2 | 0 | 0 | 0 | 0 | 0 | 10 | 191 | 7 | 0 |
| Armigeres subalbatus | 5 | 40 | 0 | 0 | 105 | 0 | 0 | 0 | 66 | 216 | 14 | 0 |
| Coquillettidia crassipes | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 |
| Culex annulus | 4 | 606 | 22 | 7 | 52 | 2 | 645 | 15 | 472 | 1825 | 50 | 1 * |
| Culex fuscocephala | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 12 | 14 | 3 | 0 |
| Culex pipiens form molestus | 63 | 19 | 53 | 0 | 87 | 0 | 17 | 0 | 21 | 260 | 21 | 0 |
| Culex quinquefasciatus | 15 | 11 | 29 | 0 | 174 | 7 | 116 | 0 | 133 | 485 | 25 | 0 |
| Culex sitiens | 0 | 0 | 0 | 0 | 62 | 0 | 0 | 0 | 4 | 66 | 5 | 0 |
| Culex tritaeniorhynchus | 2 | 2470 | 71 | 1301 | 914 | 2280 | 327 | 1473 | 2062 | 10,900 | 231 | 1* |
| Heizmannia taiwanensis | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 2 | 0 |
| Mansonia uniformis | 0 | 809 | 0 | 0 | 0 | 0 | 0 | 0 | 29 | 838 | 19 | 0 |
| Tripteroides bambusa | 0 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11 | 4 | 0 |
| Uranotaenia novobscura | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 |
| Total | 183 | 4343 | 181 | 1414 | 1434 | 2294 | 1148 | 1500 | 2877 | 15,374 | 429 | 2 |
* 50 Culex annulus and 46 Culex tritaeniorhynchus were in the two TMUV positive pools, respectively.
Table 4.
Comparison of genomic sequences between TMUV-TP1906, Sitiawan virus and other Tembusu virus strains.
Table 4.
Comparison of genomic sequences between TMUV-TP1906, Sitiawan virus and other Tembusu virus strains.
| Genomic Region (% Sequence Identity) | TMUV-TP1906 | Sitiawan Virus (JX477686) | TMUV-MM1775 (MH414569) | DTMUV-SD14 (MH748542) | DTMUVs (see Table 2) |
|---|---|---|---|---|---|
| Complete genome sequence | 10,990 nt (100) | NA # | 11,001 (91.54) | 11,001 (88.31) | 10,890–10,992 * (86.93–87.49) |
| 5′-UTR | 94 nt (100) | NA # | 94 nt (94.68) | 94 nt (92.55) | 93–95 (90.43–93.68) |
| 3′-UTR (total length) | 618 nt (100) | NA # | 629 nt (95.15) | 629 nt (92.72) | 618 nt (89.28–92.14) |
| 3′-UTR (variable region) | 87 nt(100) | NA # | 94 nt (74.71) | 94 nt (71.26) | 84–85 nt (56.96–66.25) |
| ORF | 10,278 nt (100) | 10,278 nt (93.71) | 10,278 nt (91.27) | 10,278 nt (88.00) | 10,275–10,278 nt † (86.52–87.21) |
| polyprotein | 3425 aa (100) | 3425 aa (98.92) | 3425 aa (98.57) | 3425 aa (96.64) | 3424–3425 aa (95.71–96.61) |
| C | 360 nt (100) 120 aa (100) | 360 nt (96.67) 120 aa (99.17) | 360 nt (94.71) 120 aa (96.67) | 360 nt (90.53) 120 aa (95.00) | 360 nt (88.89–90.28) 120 aa (93.33–95.00) |
| prM | 501 nt (100) 167 aa (100) | 501 nt (91.20) 167 aa (98.20) | 501 nt (90.80) 167 aa (98.80) | 501 nt (87.00) 167 aa (95.21) | 501 nt (84.40–85.40) 167 aa (94.61–96.41) |
| E | 1503 nt (100) 501 aa (100) | 1503 nt (93.68) 501 aa( 98.40) | 1503 nt (90.21) 501 aa (98.60) | 1503 nt (87.62) 501 aa (96.41) | 1503 nt (86.09–86.96) 501 aa (96.41–97.8) |
| NS1 | 1056 nt (100) 352 aa (100) | 1056 nt (93.93) 352 aa (97.73) | 1056 nt (91.86) 352 aa (98.86) | 1056 nt (89.67) 352 aa (96.31) | 1056 nt (85.13–88.35) 352 aa (90.62–94.89) |
| NS2A | 681 nt (100) 227 aa (100) | 681 nt (92.22) 227 aa (99.12) | 681 nt (89.88) 227 aa (97.8) | 681 nt (86.43) 227 aa (93.81) | 681 nt (84.14–85.61) 227 aa (90.75–92.51) |
| NS2B | 393 nt (100) 131 aa (100) | 393 nt (91.09) 131 aa (96.95) | 393 nt (89.57) 131 aa (96.95) | 393 nt (87.28) 131 aa (93.13) | 393 nt (85.24–86.77) 131 aa (90.84–93.89) |
| NS3 | 1857 nt (100) 619 aa (100) | 1857 nt (94.18) 619 aa (99.52) | 1857 nt (91.76) 619 aa (99.03) | 1857 nt (89.01) 619 aa (98.71) | 1857 nt (87.51–88.26) 619 aa (97.58–98.71) |
| NS4A | 378 nt (100) 126 aa (100) | 378 nt (93.39) 126 aa (99.21) | 378 nt (90.74) 126 aa (98.41) | 378 nt (86.21) 126 aa (96.03) | 378 nt (84.62–87.04) 126 aa (92.86–96.03) |
| 2K | 69 nt (100) 23 aa (100) | 69 nt (95.65) 23 aa (100) | 69 nt (94.12) 23 aa (100) | 69 nt (94.12) 23 aa (95.65) | 69 nt (85.51–91.18) 23 aa (95.65) |
| NS4B | 762 nt (100) 254 aa (100) | 762 nt (93.83) 254 aa (99.61) | 762 nt (90.94) 254 aa (99.21) | 762 nt (85.9) 254 aa (95.28) | 762 nt (84.83–88.52) 254 aa (91.34–97.64) |
| NS5 | 2715 nt (100) 905 aa (100) | 2715 nt (94.18) 905 aa (99.34) | 2715 nt (91.71) 905 aa (98.56) | 2715 nt (88.32) 905 aa (97.79) | 2712–2715 nt † (87.18–87.81) 904–905 aa † (96.91–98.34) |
# NA: Not available. Sequence is not available in GenBank database; * 5 DTMUV strains including DK/TH/CU-1, BYD-1, CK-SD-11, DK/TH/CU-DTMUV and G23 which do not deposit complete genome sequences in GenBank were not analyzed; † NS5 protein of both DTMUV D1977 and D1921 strains composed of 904 amino acids
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Peng, S.-H.; Su, C.-L.; Chang, M.-C.; Hu, H.-C.; Yang, S.-L.; Shu, P.-Y. Genome Analysis of a Novel Tembusu Virus in Taiwan. Viruses 2020, 12, 567. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050567
AMA Style
Peng S-H, Su C-L, Chang M-C, Hu H-C, Yang S-L, Shu P-Y. Genome Analysis of a Novel Tembusu Virus in Taiwan. Viruses. 2020; 12(5):567. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050567
Chicago/Turabian StylePeng, Shih-Huan, Chien-Ling Su, Mei-Chun Chang, Huai-Chin Hu, Su-Lin Yang, and Pei-Yun Shu. 2020. "Genome Analysis of a Novel Tembusu Virus in Taiwan" Viruses 12, no. 5: 567. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050567
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.