The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections

 , , , ,
on behalf of the VIZIONS Consortium
, , , ,
on behalf of the VIZIONS Consortium
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Ethical Approvals
2.2. The Sentinel Cohort Study and Samples
2.3. Metagenomic Next-Generation Sequencing (MNGs) Assay
2.4. Analysis of mNGS Sequence Data
2.5. PCR Confirmatory Testing of Viruses Detected by Metagenomics and Genome Sequencing
2.6. PCR Screening by New Primers Designed Based on mNGS Contigs
2.7. Viral Genotyping
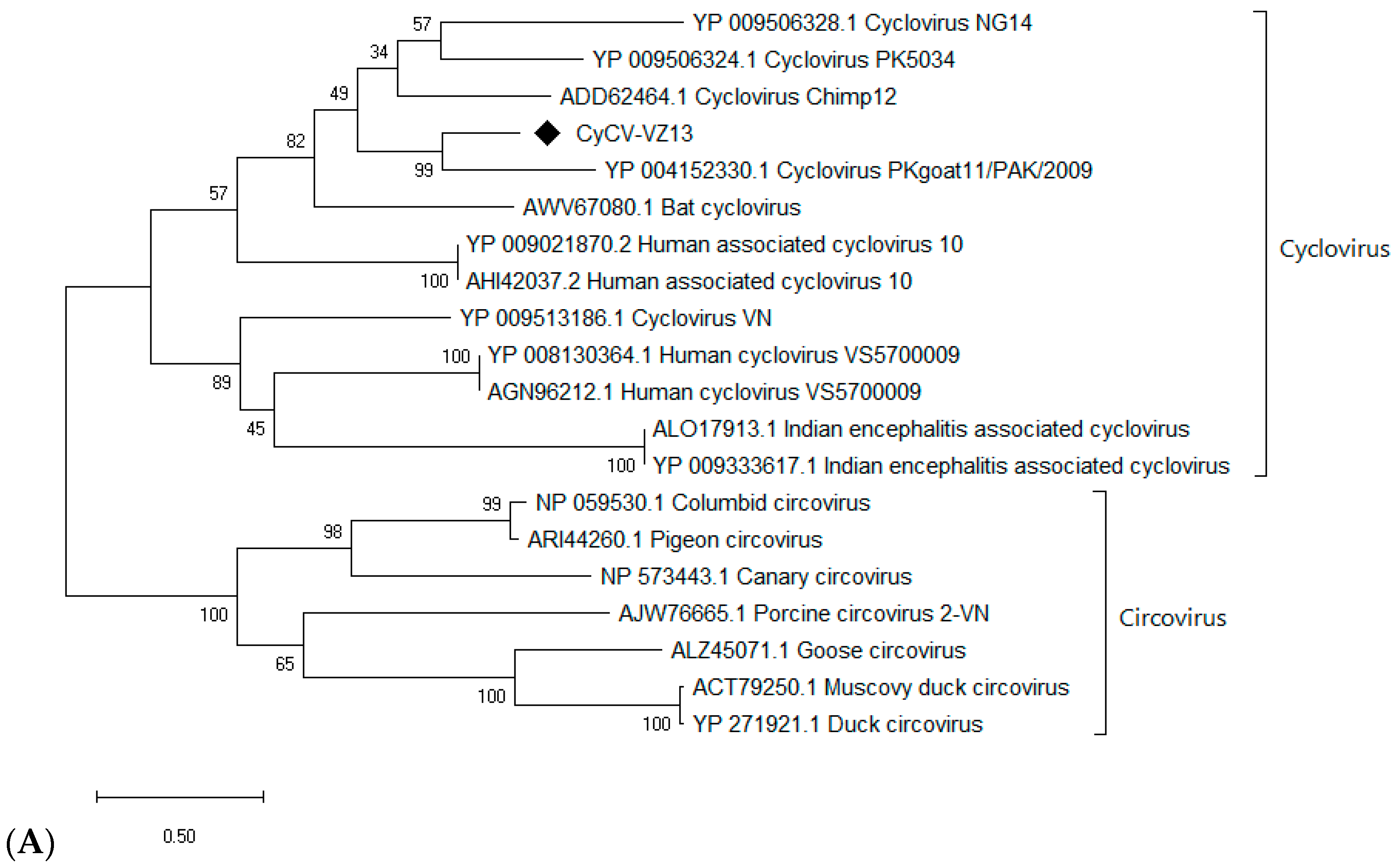
2.8. Phylogenetic Analysis
2.9. Nucleotide Sequence Accession Numbers
2.10. Statistics
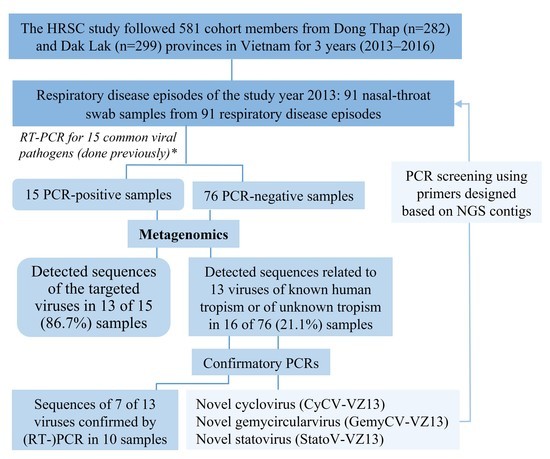
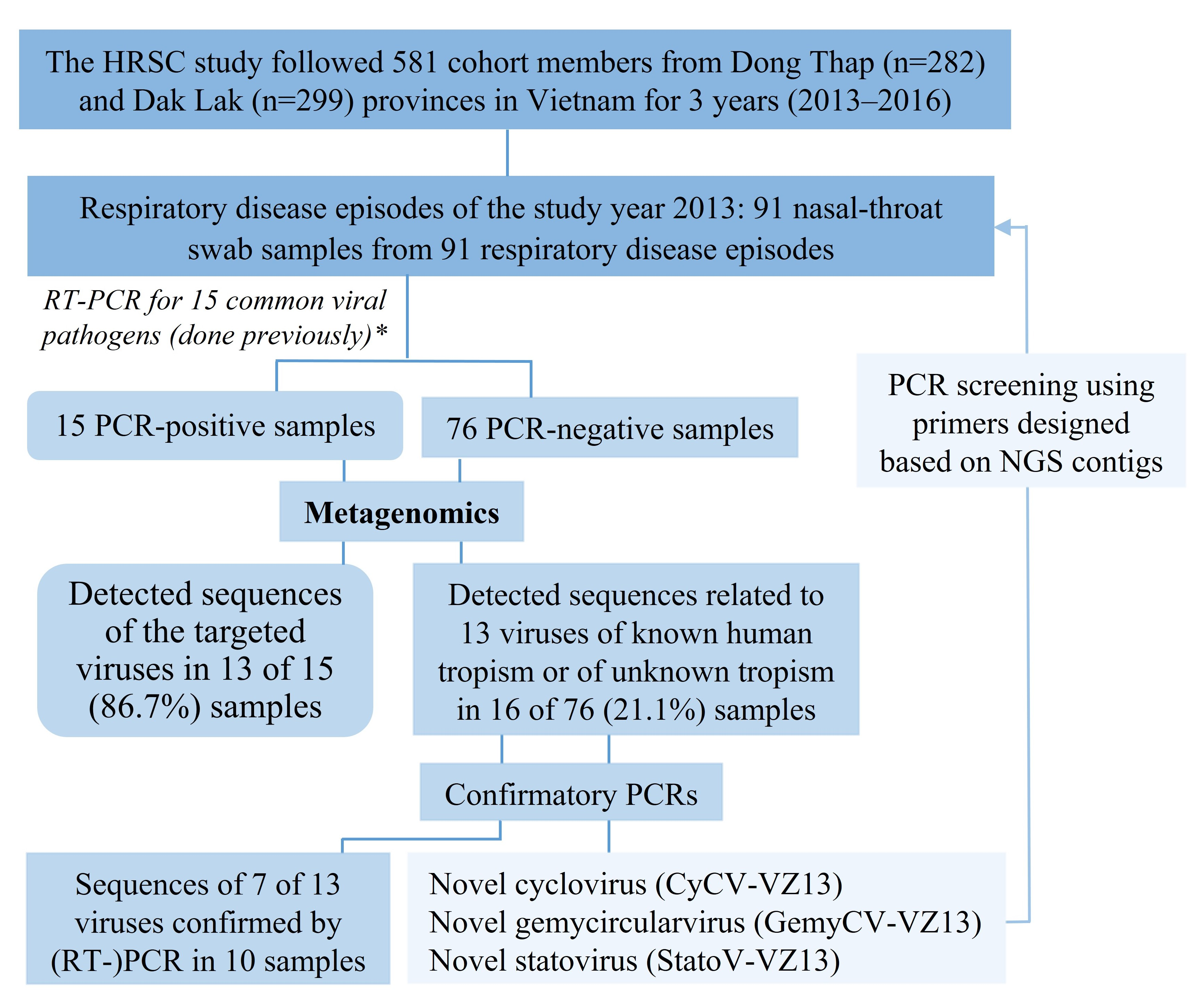
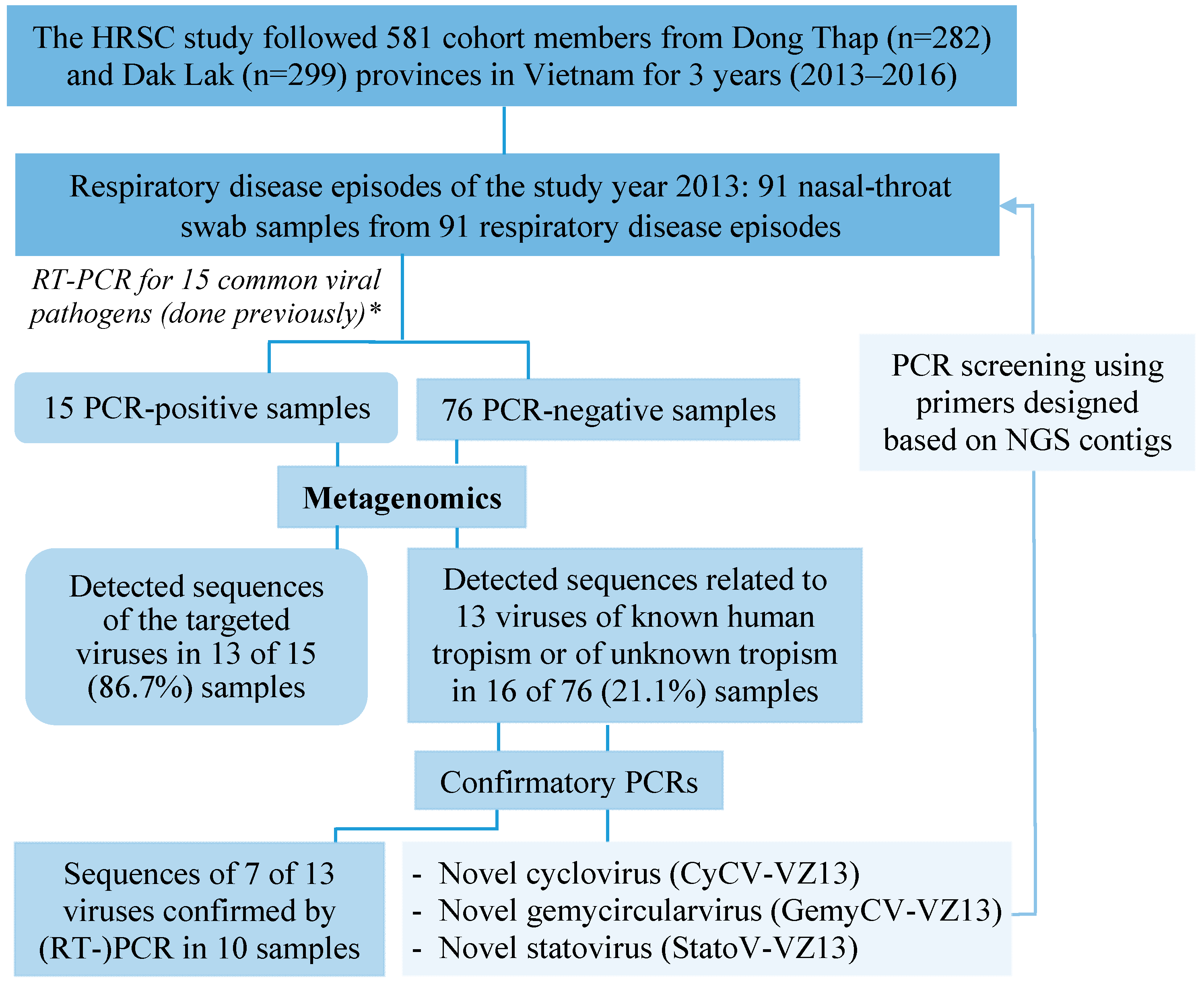
3. Results
3.1. Characteristics of the Cohort Members and Clinical Samples
3.2. Overview of Sequences Detected by Metagenomics
3.3. Viral Detection in Positive Controls
3.4. Viral Detection in RT-PCR-Negative Swabs and Results of Confirmatory PCR
3.5. Detection and Genomic Characterization of Novel Viruses
3.5.1. A Novel Cyclovirus
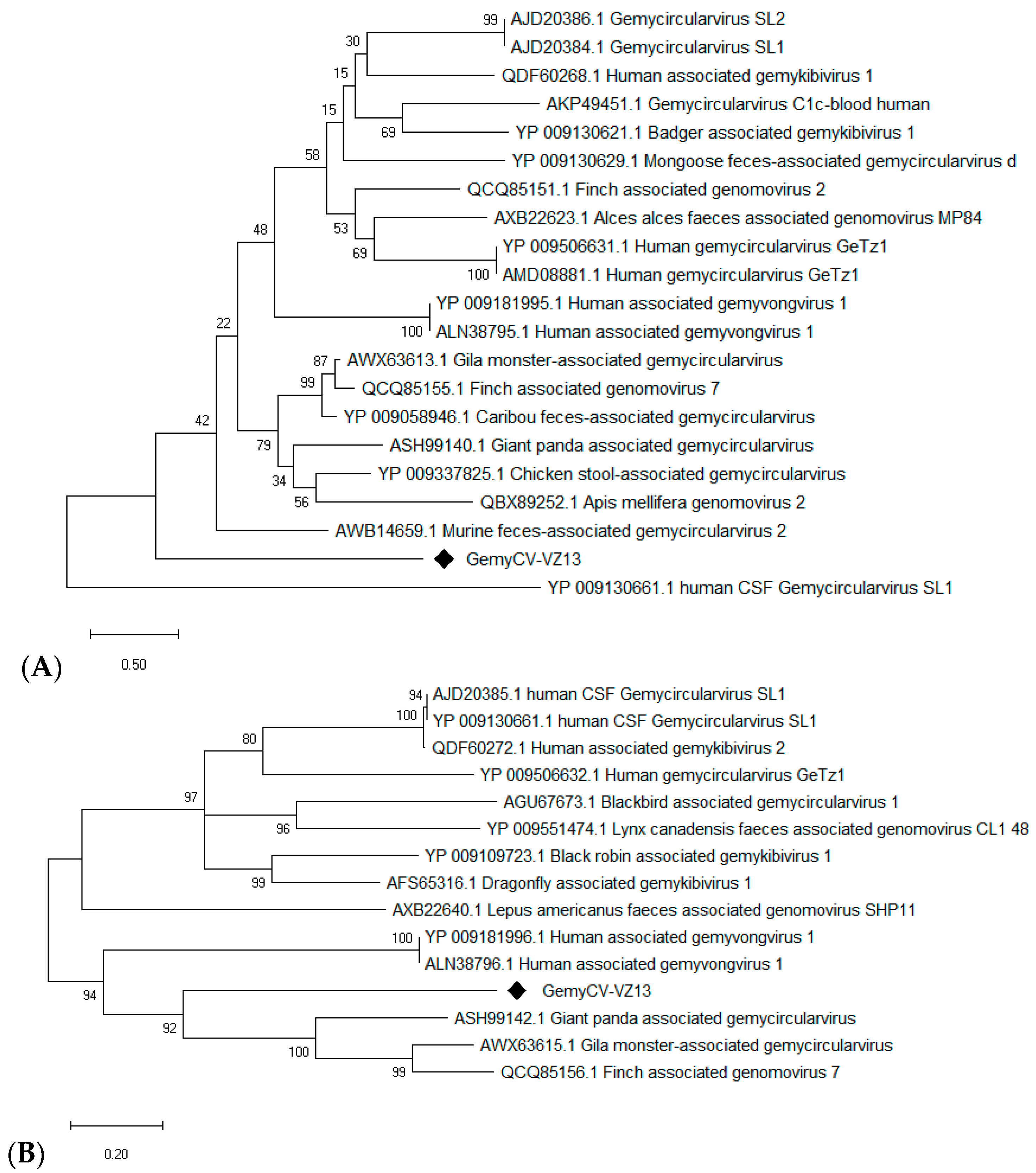
3.5.2. A Novel Gemycircularvirus
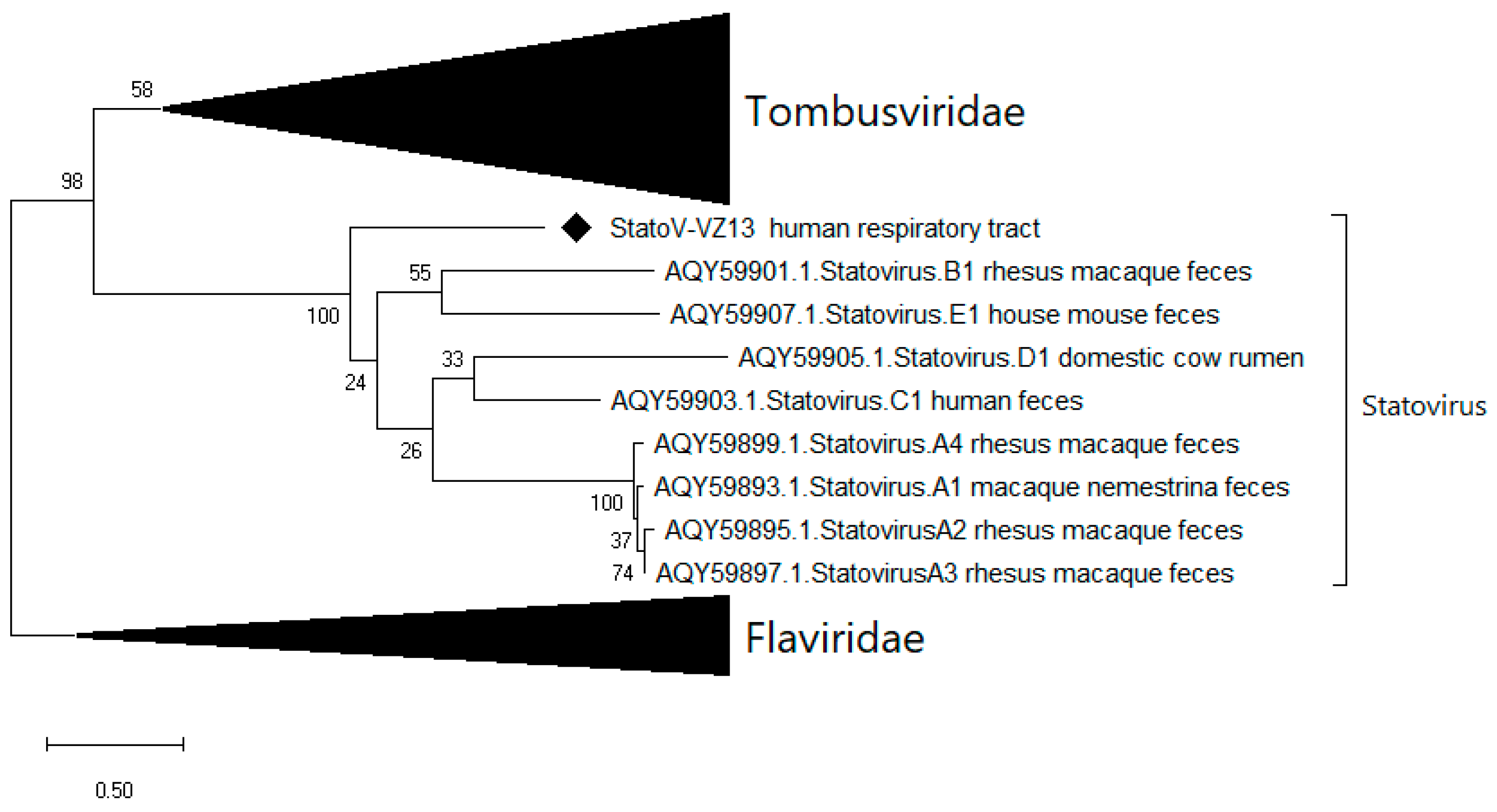
3.5.3. A Novel Statovirus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Research Needs for the Battle Against Respiratory Viruses (BRaVe); WHO: Geneva, Switzerland, 2013; pp. 1–35. [Google Scholar]
- Forum of International Respiratory Societies. The Global Impact of Respiratory Disease, 2nd ed.; European Respiratory Society: Sheffield, UK, 2017. [Google Scholar]
- Prasetyo, A.A.; Desyardi, M.N.; Tanamas, J.; Suradi; Reviono; Harsini; Kageyama, S.; Chikumi, H.; Shimizu, E. Respiratory Viruses And Torque Teno Virus in Adults With Acute Respiratory Infections. Intervirology 2015, 58, 57–68. [Google Scholar] [CrossRef]
- Vong, S.; Guillard, B.; Borand, L.; Rammaert, B.; Goyet, S.; Te, V.; Try, P.L.; Hem, S.; Rith, S.; Ly, S.; et al. Acute Lower Respiratory Infections in ≥5 Year -Old Hospitalized Patients in Cambodia, A Low-Income Tropical Country: Clinical Characteristics And Pathogenic Etiology. BMC Infect. Dis. 2013, 13, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.K.; Ngo, T.T.; Tran, P.M.; Pham, T.T.T.; Vu, H.T.T.; Nguyen, N.T.H.; Thwaites, G.; Virtala, A.-M.K.; Vapalahti, O.; Baker, S.; et al. Respiratory Viruses in Individuals With A High Frequency of Animal Exposure in Southern And Highland Vietnam. J. Med. Virol. 2019, 92, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; Nguyen, H.T.T.; Le, N.N.T.; Tran, T.T.; Lau, C.-Y.; Limmathurotsakul, D.; Deng, X.; Rahman, M.; Nguyen, C.V.V.; Van Doorn, H.R.; et al. Viruses in Vietnamese Patients Presenting With Community Acquired Sepsis of Unknown Cause. J. Clin. Microbiol. 2019, 57, e00386-19. [Google Scholar] [CrossRef] [Green Version]
- Mai, N.T.H.; Phu, N.H.; Nhu, L.N.T.; Hong, N.T.T.; Hanh, N.H.H.; Nguyet, L.A.; Phuong, T.M.; Mcbride, A.; Ha, D.Q.; Nghia, H.D.T.; et al. Central Nervous System Infection Diagnosis By Next-Generation Sequencing: A Glimpse Into The Future? Open Forum Infect. Dis. 2017, 4, 2–4. [Google Scholar] [CrossRef]
- Hong, N.T.T.; Anh, N.T.; Mai, N.T.H.; Nghia, H.D.T.; Nhu, L.N.T.; Thanh, T.T.; Phu, N.H.; Deng, X.; Van Doorn, H.R.; Chau, N.V.V.; et al. Performance Of Metagenomic Next-Generation Sequencing For The Diagnosis of Viral Meningoencephalitis in A Resource-Limited Setting. Open Forum Infect. Dis. 2020, 7, ofaa046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Coronavirus Disease 2019 (Covid-19): Situation Report—51. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200311-sitrep-51-covid-19.pdf?sfvrsn=1ba62e57_10 (accessed on 19 May 2020).
- World Health Organization. Pandemic (H1n1) 2009—Update 67. Available online: https://www.who.int/csr/don/2009_09_25/en/ (accessed on 19 May 2020).
- World Health Organization. Epidemic And Pandemic-Prone Diseases: Mers Situation Update, January 2020. Available online: http://www.emro.who.int/pandemic-epidemic-diseases/mers-cov/mers-situation-update-january-2020.html (accessed on 19 May 2020).
- World Health Organization. Sars (Severe Acute Respiratory Syndrome). Available online: https://www.who.int/ith/diseases/sars/en/ (accessed on 19 May 2020).
- World Health Organization. Zoonoses—Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/zoonoses (accessed on 10 November 2019).
- Tu, N.T.K.; Tue, N.T.; Vapalahti, O.; Virtala, A.-M.K.; Van Tan, L.; Rabaa, M.A.; Carrique-Mas, J.; Thwaites, G.E.; Baker, S. Occupational Animal Contact in Southern And Central Vietnam. Ecohealth 2019, 16, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Rabaa, M.A.; Tue, N.T.; Phuc, T.M.; Carrique-Mas, J.; Saylors, K.; Cotten, M.; Bryant, J.E.; Dang, H.; Nghia, T.; Van Cuong, N.; et al. The Vietnam Initiative On Zoonotic Infections (Vizions): A Strategic Approach To Studying Emerging Zoonotic Infectious Diseases. Ecohealth 2015, 12, 726–735. [Google Scholar] [CrossRef] [Green Version]
- Carrique-Mas, J.J.; Tue, N.T.; Bryant, J.E.; Saylors, K.; Cuong, N.V.; Hoa, N.T.; An, N.N.; Hien, V.B.; Lao, P.V.; Tu, N.C.; et al. The Baseline Characteristics And Interim Analyses of The High-Risk Sentinel Cohort of The Vietnam Initiative On Zoonotic Infections (Vizions). Sci. Rep. 2015, 5, 17965. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Tran, T.T.; Hoang, V.M.T.; Nghiem, N.M.; Le, N.N.T.; Le, T.T.M.; Phan, Q.T.; Truong, K.H.; Le, N.N.T.; Ho, V.L.; et al. Development And Evaluation of A Non-Ribosomal Random Pcr And Next-Generation Sequencing Based Assay For Detection And Sequencing of Hand, Foot And Mouth Disease Pathogens. Virol. J. 2016, 13, 125. [Google Scholar] [CrossRef] [Green Version]
- Altan, E.; Delaney, M.A.; Colegrove, K.M.; Spraker, T.R.; Wheeler, E.A.; Deng, X.; Li, Y.; Gulland, F.M.D.; Delwart, E. Complex Virome in A Mesenteric Lymph Node From A Californian Sea Lion (Zalophus Californianus) With Polyserositis And Steatitis. Viruses 2020, 12, 793. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An Ensemble Strategy That Significantly Improves De Novo Assembly of Microbial Genomes From Metagenomic Next-Generation Sequencing Data. Nucleic Acids Res. 2015, 43, E46. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast And Sensitive Protein Alignment Using Diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An Automated Genotyping Tool For Enteroviruses And Noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Coronaviridae. ICTV 9th Report (2011). Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/222/coronaviridae (accessed on 16 May 2020).
- International Committee on Taxonomy of Viruses (ICTV). Genus: Orthopneumovirus. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/negative-sense-rna-viruses/mononegavirales/w/pneumoviridae/738/genus-orthopneumovirus (accessed on 16 May 2020).
- International Committee on Taxonomy of Viruses (ICTV). ICTV 9th Report: Orthomyxoviridae Family. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/negative-sense-rna-viruses-2011/w/negrna_viruses/209/orthomyxoviridae (accessed on 16 May 2020).
- Benjamini, Y.; Hochberg, Y. Controlling The False Discovery Rate: A Practical And Powerful Approach To Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Mapping S d. FDR Online Calculator. 2020. Available online: https://www.sdmproject.com/utilities/?show=FDR (accessed on 14 April 2020).
- Sergeant, E. Epitools Epidemiological Calculators. Ausvet Pty Ltd. 2019. Available online: http://epitools.ausvet.com.au. (accessed on 14 April 2020).
- Yinda, C.K.; Zell, R.; Deboutte, W.; Zeller, M.; Conceição-Neto, N.; Heylen, E.; Maes, P.; Knowles, N.J.; Ghogomu, S.M.; Van Ranst, M.; et al. Highly Diverse Population of Picornaviridae And Other Members of The Picornavirales, In Cameroonian Fruit Bats. BMC Genom. 2017, 18, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbart, M.; Delwart, E.; Rosario, K.; Segalés, J.; Varsani, A. ICTV Virus Taxonomy Profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A. Establishing Eight New Genera And Seventy Three Species in The Family Genomoviridae. Virus Evol. 2017, 3, vew037. [Google Scholar]
- Janowski, A.B.; Krishnamurthy, S.R.; Lim, E.S.; Zhao, G.; Brenchley, J.M.; Barouch, D.H.; Thakwalakwa, C.; Manary, M.J.; Holtz, L.R.; Wang, D. Statoviruses, A Novel Taxon of Rna Viruses Present in The Gastrointestinal Tracts of Diverse Mammals. Virology 2017, 504, 36–44. [Google Scholar] [CrossRef]
- Graf, E.H.; Simmon, K.E.; Tardif, K.D.; Hymas, W.; Flygare, S.; Eilbeck, K.; Yandell, M.; Schlaberg, R. Unbiased Detection of Respiratory Viruses By Use of Rna Sequencing-Based Metagenomics: A Systematic Comparison To A Commercial Pcr Panel. J. Clin. Microbiol. 2016, 54, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y.; Miller, S.A. Clinical Metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Van Tan, L.; Van Doorn, H.R.; Nghia, H.D.T.; Chau, T.T.H.; Tu, L.T.P.; De Vries, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; Baker, S.; et al. Identification of A New Cyclovirus in Cerebrospinal Fluid of Patients With Acute Central Nervous System Infections. Mbio 2013, 4, e00231-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, D. Circoviruses: Immunosuppressive Threats to Avian Species: A Review. Avian Pathol. 2000, 29, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting The Taxonomy of the Family Circoviridae: Establishment of the Genus Cyclovirus and Removal of the Genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [Green Version]
- Sauvage, V.; Gomez, J.; Barray, A.; Vandenbogaert, M.; Boizeau, L.; Tagny, C.T.; Rakoto, O.; Bizimana, P.; Guitteye, H.; Ciré, B.B.; et al. High Prevalence of Cyclovirus Vietnam (Cycv-Vn) in Plasma Samples From Madagascan Healthy Blood Donors. Infect. Genet. Evol. 2018, 66, 9–12. [Google Scholar] [CrossRef]
- Smits, S.L.; Zijlstra, E.E.; van Hellemond, J.J.; Schapendonk, C.M.E.; Bodewes, R.; Schürch, A.C.; Haagmans, B.L.; Osterhaus, A.D.M.E. Novel cyclovirus in human cerebrospinal fluid, Malawi, 2010-2011. Emerg Infect Dis. 2013, 19, 1511–1513. [Google Scholar] [CrossRef]
- Phan, T.G.; Luchsinger, V.; Avendaño, L.F.; Deng, X.; Delwart, E. Cyclovirus in nasopharyngeal aspirates of Chilean children with respiratory infections. J. Gen. Virol. 2014, 95, 922–927. [Google Scholar] [CrossRef]
- Macera, L.; Focosi, D.; Vatteroni, M.L.; Manzin, A.; Antonelli, G.; Pistello, M.; Maggi, F. Cyclovirus Vietnam Dna in Immunodeficient Patients. J. Clin. Virol. 2016, 81, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Macera, L.; Spezia, P.G.; Medici, C.; Falasca, F.; Sciandra, I.; Antonelli, G.; Focosi, D.; Pistello, M.; Maggi, F. Low Prevalence of Gemycircularvirus Dna in Immunocompetent and Immunocompromised Subjects. New Microbiol. 2019, 42, 118–120. [Google Scholar]
- Zhou, C.; Zhang, S.; Gong, Q.; Hao, A. A Novel Gemycircularvirus in An Unexplained Case of Child Encephalitis. Virol. J. 2015, 12, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.G.; Mori, D.; Deng, X.; Rajidrajith, S.; Ranawaka, U.; Fei, T.; Ng, F.; Bucardo-Rivera, F.; Orlandi, P.; Ahmed, K.; et al. Small Viral Genomes in Unexplained Cases of Human Encephalitis, Diarrhea, and in Untreated Sewage. Virology 2015, 482, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberto, I.; Gunst, K.; Müller, H.; Zur Hausen, H.; De Villiers, E.M. Mycovirus-Like Dna Virus Sequences From Cattle Serum and Human Brain and Serum Samples From Multiple Sclerosis Patients. Genome Announc. 2014, 2, 2014. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, L.; Deng, X.; Blümel, J.; Nübling, C.M.; Hunfeld, A.; Baylis, S.A.; Delwart, E. Viral Nucleic Acids in Human Plasma Pools. Transfusion 2016, 56, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating Viral Sequences in High-Throughput Sequencing Viromics: A Linkage Study of 700 Sequencing Libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielian, M.; Mettenleiter, T.C.; Roossinck, M.J. Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2019; Chapter 3; pp. 71–133. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mNGS Analysis | ||||
|---|---|---|---|---|
| Total | PCR Positive | PCR Negative | p Value #^ | |
| No. of cohort members | N = 58 | N = 14 | N = 51 | |
| Median age (range) (in years) | 35.5 (7–76) | 31 (13–58) | 38 (7–76) | 0.465 ## |
| Sex ratio (male/female) | 2.6 (42/16) | 1.3 (8/6) | 3.3 (39/12) | 0.465 |
| Occupations | ||||
| Animal health worker | 12 (20.7) | 2 (14.3) | 11 (21.6) | 1 |
| Animal-raising farmer | 26 (44.8) | 6 (42.9) | 22 (43.1) | 1 |
| Slaughterer | 18 (31.0) | 5 (35.7) | 17 (33.3) | 1 |
| Rat-trader | 2 (3.4) | 1 (7.1) | 1 (2.0) | 0.829 |
| Having chronic diseases | 4 (6.9) | 0 | 4 (7.8) | 1 |
| Respiratory disease episodes | N = 91 | N = 15 | N = 76 | |
| Frequency of clinical signs | ||||
| Fever | 91 (100) | 15 (100) | 76 (100) | - |
| Cough | 75 (82.4) | 8 (53.3) | 67 (88.2) | 0.015 |
| Sneezing | 69 (75.8) | 14 (93.3) | 55 (72.4) | 0.465 |
| Sore throat | 49 (53.8) | 8 (53.3) | 41 (53.9) | 1 |
| Dyspnea | 9 (9.9) | 1 (6.7) | 8 (10.5) | 1 |
| Headache | 57 (62.6) | 12 (80) | 45 (59.2) | 0.465 |
| Body aches | 47 (51.6) | 7 (46.7) | 40 (52.6) | 1 |
| Watery diarrhea | 11 (12.1) | 0 (0) | 11 (14.5) | 0.5 |
| Nausea | 2 (2.2) | 0 (0) | 2 (2.6) | 1 |
| No. | Multiplex RT-PCR ** | NGS Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Sample ID | Virus Detected | Ct Value | Virus Genotype | Reads (%) # | Total Length (bp) | Genome Coverage (%) | Other Virus Detected ## | |
| 1 | 72 | EVs | 32.4 | Coxsackievirus A21 | 52,989 (12) | 7440 | 100.0 | |
| HRV | 37.1 | HRV C56 | 2506 (0.6) | 7099 | 98.1 | |||
| 2 | 75 | EVs | 38.6 | HRV B | 4 (0.0) | 598 | 8.3 | |
| 3 | 5 | HRV | 38.4 | HRV B3 | 678 (0.7) | 5512 | 75.0 | Human betaherpesvirus 7 |
| 4 | 33 | HRV | 40 | EVs-D68 | 3174 (0.7) | 5629 | 76.2 | |
| 5 | 54 | HRV | 40 | HRV B | 6 (0.0) | 723 | 10.0 | |
| 6 | 73 | HRV | 40 | HRV B86 | 6644 (1.5) | 7212 | 99.2 | Vientovirus |
| 7 | 83 | HRV | 38.7 | HRV B79 | 6157 (1.8) | 5639 | 78.2 | Novel gemycircularvirus (GemyCV-VZ13) |
| 8 | 86 | HRV | 38.2 | HRV B79 | 19,606 (5.6) | 7224 | 99.7 | |
| 9 | 91 | HRV | 40 | HRV A57 | 2538 (1.1) | 3450 | 47.8 | |
| 10 | 92 | HRV | 36.5 | HRV B35 | 12,481 (3.1) | 7298 | 100.0 | Bat badicivirus, bat posalivirus |
| 11 | 4 | Influenza A virus | 29.3 | Influenza A/N2 virus | 2 (0.0) | 115 | 0.8 | |
| 12 | 6 | CoV * | 36 | CoV OC43 | 8 (0.0) | 733 | 2.4 | |
| 13 | 52 | RSV-A | 30.8 | RSV-A genotype ON1 | 236 (0.1) | 5398 | 35.4 | |
| 14 | 39 | RSV-A | 36.3 | Not detected | 0 | 0 | 0 | |
| 15 | 65 | MPV | 39.5 | Not detected | 0 | 0 | 0 | |
| No. | Sample ID | Detected Viruses Previously Reported in Human Samples | Confirmed by PCR | No. of Reads | Total Contig Length (bp) | Amino Acid Identity to GenBank Strain (%) | Genome Coverage (%) |
|---|---|---|---|---|---|---|---|
| 1 | 89 | Rotavirus | Yes | 17 | 360 | 98 | 1.9 |
| 2 | 73 | Vientovirus *# | Yes | 2 | 146 | 53 | 4.8 |
| 3 | 23 | Novel cyclovirus (CyCV-VZ13) | Yes | 5 | 448 | 61.8 | 25.9 |
| 4 | 32 | Novel gemycircularvirus virus (GemyCV-VZ13) | Yes | 1852 | 1995 | 39 | 91 |
| 5 | 83 | GemyCV-VZ13 | Yes | 120 | 2000 | 45 | 92 |
| 6 | 89 | GemyCV-VZ13 | Yes | 1 | 148 | 46.9 | 6.8 |
| 7 | 24 | Novel statovirus (StatoV-VZ13) | Yes | 91 | 1018 | 42.5 | 24.6 |
| 8 | 32 | StatoV-VZ13 | Yes | 5 | 231 | 35 | 5.6 |
| 9 | 82 | StatoV-VZ13 | Yes | 27 | 2000 | 49 | 48.4 |
| 10 | 87 | Gemycircularvirus | Yes | 39 | 858 | 83 | 39 |
| 11 | 71 | Gemycircularvirus | Yes | 117 | 1400 | 97 | 63.7 |
| 12 | 88 | Gemycircularvirus | Yes | 2 | 300 | 73 | 13.6 |
| 13 | 11 | Statovirus | Yes | 4 | 351 | 91 | 8.5 |
| 14 | 71 | Statovirus | Yes | 7 | 812 | 90 | 19.6 |
| 15 | 5 | Human betaherpesvirus 7 * | Not done | 2 | 295 | 100 | 0.2 |
| 16 | 15 | Human papillomavirus | Not done | 73 | 1280 | 99.3 | 17.5 |
| 17 | 17 | Human papillomavirus | Not done | 6 | 437 | 97.9 | 6 |
| 18 | 2 | Torque teno virus | Not done | 4 | 554 | 88.4 | 14.6 |
| 19 | 68 | Torque teno virus | Not done | 2 | 217 | 70.6 | 5.7 |
| 20 | 24 | MPV | No | 6 | 417 | 100 | 3.1 |
| 21 | 47 | RSV A | No | 6 | 468 | 100 | 3.1 |
| 22 | 92 | Bat badicivirus-like virus * | No | 2 | 204 | 49 | 2.3 |
| 23 | 92 | Bat posalivirus-like virus * | No | 3 | 182 | 56 | 2 |
| 24 | 83 | Viruses of Circoviridae family | No | 10 | 167 | 64 | 7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thi Kha Tu, N.; Thi Thu Hong, N.; Thi Han Ny, N.; My Phuc, T.; Thi Thanh Tam, P.; Doorn, H.R.v.; Dang Trung Nghia, H.; Thao Huong, D.; An Han, D.; Thi Thu Ha, L.; et al. The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections. Viruses 2020, 12, 960. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090960
Thi Kha Tu N, Thi Thu Hong N, Thi Han Ny N, My Phuc T, Thi Thanh Tam P, Doorn HRv, Dang Trung Nghia H, Thao Huong D, An Han D, Thi Thu Ha L, et al. The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections. Viruses. 2020; 12(9):960. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090960
Chicago/Turabian StyleThi Kha Tu, Nguyen, Nguyen Thi Thu Hong, Nguyen Thi Han Ny, Tran My Phuc, Pham Thi Thanh Tam, H. Rogier van Doorn, Ho Dang Trung Nghia, Dang Thao Huong, Duong An Han, Luu Thi Thu Ha, and et al. 2020. "The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections" Viruses 12, no. 9: 960. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090960