Merkel Cell Polyomavirus in Merkel Cell Carcinoma: Integration Sites and Involvement of the KMT2D Tumor Suppressor Gene


Abstract
:1. Introduction
2. Methods
2.1. MCPyV Integration Analysis
2.2. PCR
2.3. MCPyV Integration Site Meta-Analysis
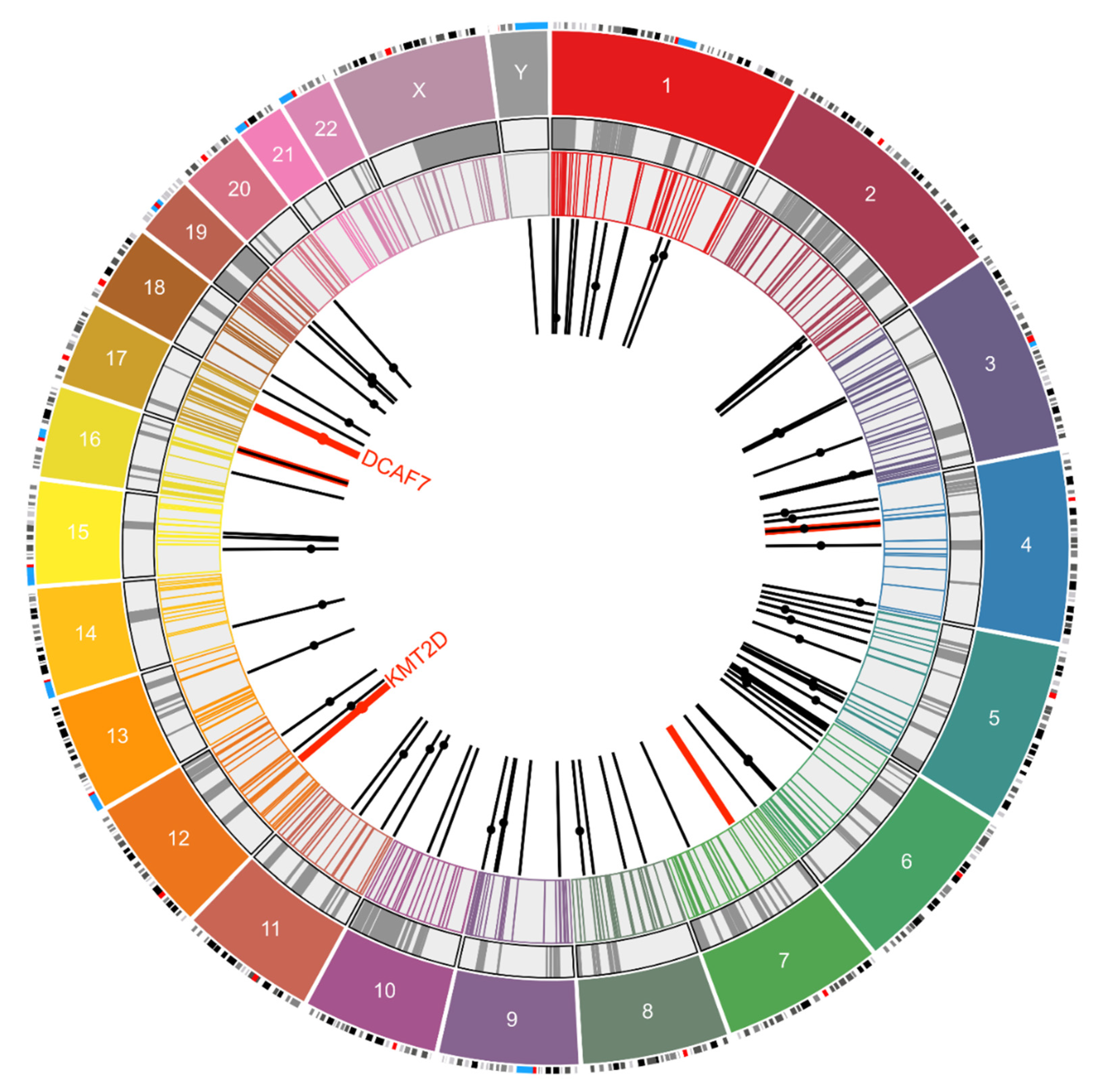
3. Result
3.1. Integration Site Detection from Exome Sequencing Data
3.2. Correlation with Previously Reported MCPyV Integration Sites
3.3. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, W.; Huang, Y.-J.; Liu, C.; Yang, Y.; Liu, H.; Cui, J.-G.; Cheng, Y.; Gao, F.; Cai, J.-M.; Li, B.-L. Inhibition of TBK1 attenuates radiation-induced epithelial–mesenchymal transition of A549 human lung cancer cells via activation of GSK-3β and repression of ZEB1. Lab. Investig. 2014, 94, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, K.L.; Healy, M.A.; Nghiem, P.T.; Sober, A.J.; Johnson, T.M.; Bichakjian, C.K.; Wong, S.L. Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann. Surg. Oncol. 2016, 23, 3564–3571. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.T.; Wong, M.K.K.; Brownell, I. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Chang, Y.; Moore, P.S. MCV and Merkel cell carcinoma: A molecular success story. Curr. Opin. Virol. 2012, 2, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.; Becker, J.C. Merkel Cell Polyomavirus-Infected Merkel Cell Carcinoma Cells Require Expression of Viral T Antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.-M.; Han, S.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8, e02079-16. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.; Chang, Y. Common Commensal Cancer Viruses. PLoS Pathog. 2017, 13, e1006078. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Schrama, D.; Sarosi, E.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.; Utikal, J.S.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Sastre-Garau, X.; Peter, M.; Avril, M.-F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Laude, H.C.; Jonchère, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.-F.; Dupin, N.; et al. Distinct Merkel Cell Polyomavirus Molecular Features in Tumour and Non Tumour Specimens from Patients with Merkel Cell Carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar] [CrossRef] [PubMed]
- Doolittle-Hall, J.M.; Glasspoole, D.C.; Seaman, W.T.; Webster-Cyriaque, J. Meta-Analysis of DNA Tumor-Viral Integration Site Selection Indicates a Role for Repeats, Gene Expression and Epigenetics. Cancers 2015, 7, 2217–2235. [Google Scholar] [CrossRef]
- Czech-Sioli, M.; Guenther, T.; Therre, M.; Spohn, M.; Indenbirken, D.; Theiss, J.; Riethdorf, S.; Qi, M.; Alawi, M.; Wülbeck, C.; et al. High-resolution analysis of Merkel Cell Polyomavirus in Merkel Cell Carcinoma reveals distinct integration patterns and suggests NHEJ and MMBIR as underlying mechanisms. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Leeman, J.E.; Li, Y.; Bell, A.; Hussain, S.S.; Majumdar, R.; Rong-Mullins, X.; Blecua, P.; Damerla, R.; Narang, H.; Ravindran, P.T.; et al. Human papillomavirus 16 promotes microhomology-mediated end-joining. Proc. Natl. Acad. Sci. USA 2019, 116, 21573–21579. [Google Scholar]
- Duncavage, E.J.; Magrini, V.; Becker, N.; Armstrong, J.R.; Demeter, R.T.; Wylie, T.; Abel, H.J.; Pfeifer, J.D. Hybrid Capture and Next-Generation Sequencing Identify Viral Integration Sites from Formalin-Fixed, Paraffin-Embedded Tissue. J. Mol. Diagn. 2011, 13, 325–333. [Google Scholar] [CrossRef]
- Martel-Jantin, C.; Filippone, C.; Cassar, O.; Peter, M.; Tomasic, G.; Vielh, P.; Brière, J.; Petrella, T.; Aubriot-Lorton, M.; Mortier, L.; et al. Genetic variability and integration of Merkel cell polyomavirus in Merkel cell carcinoma. Virol. 2012, 426, 134–142. [Google Scholar] [CrossRef]
- Levin, M.K.; Wollison, B.M.; Powers, W.; Burns, R.T.; Patel, N.; Ducar, M.D.; Starrett, G.J.; Garcia, E.P.; Manning, D.K.; Cheng, J.; et al. ViroPanel: Hybrid Capture and Massively Parallel Sequencing for Simultaneous Detection and Profiling of Oncogenic Virus Infection and Tumor Genome. J. Mol. Diagn. 2020, 22, 476–487. [Google Scholar]
- García-Mulero, S.; Moratalla-Navarro, F.; Curiel-Olmo, S.; Moreno, V.; Vaque, J.P.; Sanz-Pamplona, R.; Piulats, J.M. Detection of Merkel cell polyomavirus using whole exome sequencing data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chandrani, P.; Kulkarni, V.; Iyer, P.; Upadhyay, P.; Chaubal, R.; Das, P.; Mulherkar, R.; Singh, R.; Dutt, A. NGS-based approach to determine the presence of HPV and their sites of integration in human cancer genome. Br. J. Cancer 2015, 112, 1958–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Kumar, R.; Nagpal, G.; Kumar, V.; Usmani, S.S.; Agrawal, P.; Raghava, G.P.S. HumCFS: A database of fragile sites in human chromosomes. BMC Genom. 2019, 19, 985. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Chen, L.H.; Huang, Y.; Chang, C.-C.; Wang, P.; Pirozzi, C.J.; Qin, X.; Bao, X.; Greer, P.K.; McLendon, R.E.; et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget 2013, 4, 2144–2153. [Google Scholar] [CrossRef] [Green Version]
- AACR Project GENIE Consortium. The AACR Project GENIE Consortium AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.; Lee, E.; Yue, Y.; Vandergriff, T.; Byekova, G.; Cockerell, C.; Hosler, G.; Pastrana, D.V.; Buck, C.; Wang, R. 569 Human polyomavirus 6 and 7 are associated with pruritic and dyskeratotic dermatoses. J. Investig. Dermatol. 2017, 137, S98. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.-M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389–1406. [Google Scholar] [PubMed]
- Liu, X.; Xu, Y.; Han, L.; Yi, Y. Reassessing the Potential of Myb-targeted Anti-cancer Therapy. J. Cancer 2018, 9, 1259–1266. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Year | First Author (Journal) | Detection Assay | Integration Analysis Tool | Number of MCC—Integration Sites Detected | Human Genome Integration Junction, Chromosome (# of Cases) | MCPyV Genome Integration Junction (# of Cases) |
|---|---|---|---|---|---|---|
| 2008 | Feng H. (Science) | cDNA pyrosequencing, 3’-RACE, 5’-RACE, Southern Hybridization | Digital Transcriptome Subtraction | 2 | chr3 | LT (1) |
| 2009 | Sastre-Garau X. (J Pathol) | DIPS-PCR | N/A | 10 | chr2, 3, 4, 5, 6, 8, 12, 20, and Y | LT (7), NCCR (2), VP1 (2) |
| 2010 | Laude H.C. (PLoS Pathogens) | DIPS-PCR | N/A | 4 | chr6, 9, 11, 15 | LT (3), VP1 (1) |
| 2011 | Duncavage E.J. (J Mol Diagn) | MCPyV-specific Hybrid Capture NGS | SLOPE and Breakdancer | 4 | chr6, 8, 9 | Not reported |
| 2012 | Martel-Jantin C. (Virology) | DIPS-PCR | N/A | 19 | chr1 (3),3 (1),4 (3), 5 (4), 6. (2), 14 (1), 18 (1) and 19 (2) | LT (13), ST (1), VP1(3), VP2 (2) |
| 2013 | Guastafierro A. (J Virol Methods) | Lambda phage library, 3’RACE, 5’RACE and Southern hybridization Library | N/A | 4 | chr3, 5, 6, 10 | LT (3), ST (1) |
| 2015 | Doolittle-Hall et al., Cancers | N/A (Meta-analysis of published reports) | Meta-analysis of published reports | 37 | Multiple, Non-recurrent | Not reported |
| 2017 | Starrett G.J. (mBio) | Whole Genome NGS | Custom Pipeline | 4 | chr1, 6, 19, 20 | LT (2), NCCR (2) |
| 2019 | Schrama D. et al., (Int J Cancer) | DIPS-PCR | N/A | 5 | chrs 1,2,6,11,12 | LT (2), VP1 (4), VP2 (3) |
| 2020 | Slevin M.K. (J Mol Diagn) | Hybrid-capture and Massively parallel sequencing (Viropanel) | SvABA (structural variation and insertion/deletion analysis by assembly) | 33 | chr1 (5), 2 (2), 3 (1), 5 (7), 6 (4), 7 (2), 8 (2), 9 (2), 10 (1), 11 (4), 15 (1) and 16 (2) | LT (5), ST (2), VP1 (3), VP2 (1) and ** (1) |
| 2020 (bioRxiv) | Czech-Sioli M. (bioRxiv) | Long-read third generation NGS, Nanochannel and Nanopore sequencing | Multiple | 9 | chr3, 4, 5 (4), 9, 11,13,20 | LT (5), NCCR (2), VP1 (8), VP2 (4) and ** (1) |
| 2020 (bioRxiv) | Garcia-Mulero S. (bioRxiv) | Human coding region sequencing NGS (Hybrid Capture) | No specialized analysis tool | 1 | chr 19 | Not reported |
| 2020 | Starrett G.J. (Genome Med) | Hybrid-capture and Massively parallel sequencing (Viropanel) | Custom pipeline (oncovirus tools- suite - (https://github.com/gstarrett/oncovirus_tools)) | 25 | chr1 (4), 2 (2), 3 (1), 5 (4), 6 (3), 7 (2), 8 (2), 9 (3), 10 (1), 11 (1), 15 (1), 16 (2) and 18 (1) | Not reported |
| 2020 | Arora R. (current study) | whole exome (Hybrid Capture) | HPVDetector | 5 | chr4, 7, 12, 16 and 17 | LT, NCCR (2), VP1 (2) |
| Breakpoints in the Human Genome | ||
|---|---|---|
| Chromosome | Number of Sites | Genes Associated |
| 1 | 14 | PLA2G4A, RABGAP1L, MCOLN2, CASZ1 (I#1) |
| 2 | 6 | SF3B1 (I#1) |
| 3 | 7 | PTPRG (I#1), ATP11B, EEFSEC, ADAMTS9 (I) |
| 4 | 6 | SRD5A2L2, KCNIP4, RELL1, GRID2, FRG1(I), |
| 5 | 21 | TLX3, XRCC4, ISL1, PRLR, CAMK2A(I), CD74(I) |
| 6 | 15 | IL20RA, GMDS, MAP3K7IP2, CDKAL1 (I#4), MRDS1 (I#6), CDKAL1, RP3348–I23.2 |
| 7 | 5 | |
| 8 | 6 | MYC |
| 9 | 8 | DENND1A;mir601, INPP5E (I) |
| 10 | 3 | |
| 11 | 8 | PARVA;TEAD1, LUZP2 (I#10;Tr#1), TTC9C (E#1), AHNAK (I) |
| 12 | 3 | AX747640, BEST3 (I#4;Tr#1), KMT2D (I#34) |
| 13 | 1 | DACH1 (I) |
| 14 | 1 | NOVA1 |
| 15 | 3 | ATPBD4;XP_002343377 |
| 16 | 5 | |
| 17 | 1 | DCAF7 (I#3) |
| 18 | 2 | CDH2 |
| 19 | 4 | EVI5L, ECH1, GRWD1 |
| 20 | 3 | SNTA1, CBFA2T2, BCL2L1 (E), TPX2 (I) |
| Y | 1 | |
| Total | 123 | |
| Breakpoints in the Viral Genome | ||
| Genomic Region | Number of Sites | Gene |
| Large T Antigen | 42 | LT |
| Small T Antigen | 4 | ST |
| NCCR | 8 | |
| Viral Capsid Protein 1 | 23 | VP1 |
| Viral Capsid Protein 2 | 10 | VP2 |
| Between LT and VP1 | 2 | |
| Total | 89 | |
| MCPyV Genome | |||||
|---|---|---|---|---|---|
| Genome Location at Breakpoint | Gene | Forward Read Sequence | Genome Location at Breakpoint | Gene | Reverse Read Sequence |
| chr12_49429977 | KMT2D (*I#34) | CACTTACTTCACTCGAAACAGTTTACTCCAGCCTAACCAATTCACTCACTGTTCTTGAAACATCCCATAGGTTTTCCCACCATTGCAG | MCV_DysKA_3082 | Large T antigen | AGGGCCTATTAACAGTGGAAAAACAAGCTTTGCTGCAGCCTTAATAGATTTGCTAGAAGGGAAGGCCTTGAATATAAACTGTCCATCTGATAAACTACCT |
| chr17_61656843 | DCAF7 (*I#3) | AAGAAGCCAAAATAAATTTTCTCAGATGTCACACTGGGCCATACAT CCTAGAACAAGGCTCAGTGGGAAGTAGGCTAGGCGTGGAATGTGGTTATTTATTCCTTTGTGTGGGTGAGCTTTCCGTGTGTATAGATCTTGTCTC | MCV_DysKA_172 | NCCR | GAGGCCTCGGAGGCT AGGAGCCCCAAGCCTCTGCCAACTTGAAAAAAAAAAGTCACCTAGG |
| chr4_58486053 | Intergenic | TAGATCCCTGAGGAATCGCCACACTGTCTTCCACAGTGGTTGAACTAGTATACAGTCT | MCV_DysKA_2201 | VP1 | TTATCTTTTCCTTCCATAGGTTGGCCTGACACTTTTGGCATTAAGTTGCTGAAGAGTGAGTTTGTTA |
| chr7_44780960 | Intergenic | GTTGAAAAGGGCTGAGGAAAGAAAAATAGAGAACAAAAAGGGGGTGGGTCATTTTAAATTACTT | MCV_DysKA_175 | NCCR | AGCCTCGGAGGCTAGGAGCCCCAAGCCTCTGCCAACTTGAAAAAAAAAAGTCACCTAGGC |
| chr16_85632533 | Intergenic | TGGTTGGCACAAGAAACTGCCAAGCCACAGGCCCTGCTCCATACCCTCCTCTCTCCTACTCCCCCTCAATTATTTTTCTTGGATGTTGTGAATTAAATCTTCCAGTAACCGCAGGAGACGTCTTC | MCV_DysKA_1951 | VP1 | GTCCTTTTAAATGAGAATGGAGTGGGCCCTCTATGCAAAGGAGATGGCCTATTTATTAGCTGTGCAGA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arora, R.; Choi, J.E.; Harms, P.W.; Chandrani, P. Merkel Cell Polyomavirus in Merkel Cell Carcinoma: Integration Sites and Involvement of the KMT2D Tumor Suppressor Gene. Viruses 2020, 12, 966. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090966
Arora R, Choi JE, Harms PW, Chandrani P. Merkel Cell Polyomavirus in Merkel Cell Carcinoma: Integration Sites and Involvement of the KMT2D Tumor Suppressor Gene. Viruses. 2020; 12(9):966. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090966
Chicago/Turabian StyleArora, Reety, Jae Eun Choi, Paul W. Harms, and Pratik Chandrani. 2020. "Merkel Cell Polyomavirus in Merkel Cell Carcinoma: Integration Sites and Involvement of the KMT2D Tumor Suppressor Gene" Viruses 12, no. 9: 966. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090966