
Phylogenomic Analysis of Human Papillomavirus Type 31 and Cervical Carcinogenesis: A Study of 2093 Viral Genomes

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.1.1. PaP Study
2.1.2. IARC Study
2.2. DNA Isolation, Library Construction and Next-Generation Sequencing
2.3. Viral Methylation Assay
2.4. Statistical Analyses
2.4.1. HPV31 Lineage Assignment
2.4.2. Statistical Analysis of Viral Genetic Variation
2.4.3. Statistical Analysis of Viral Methylation
3. Results
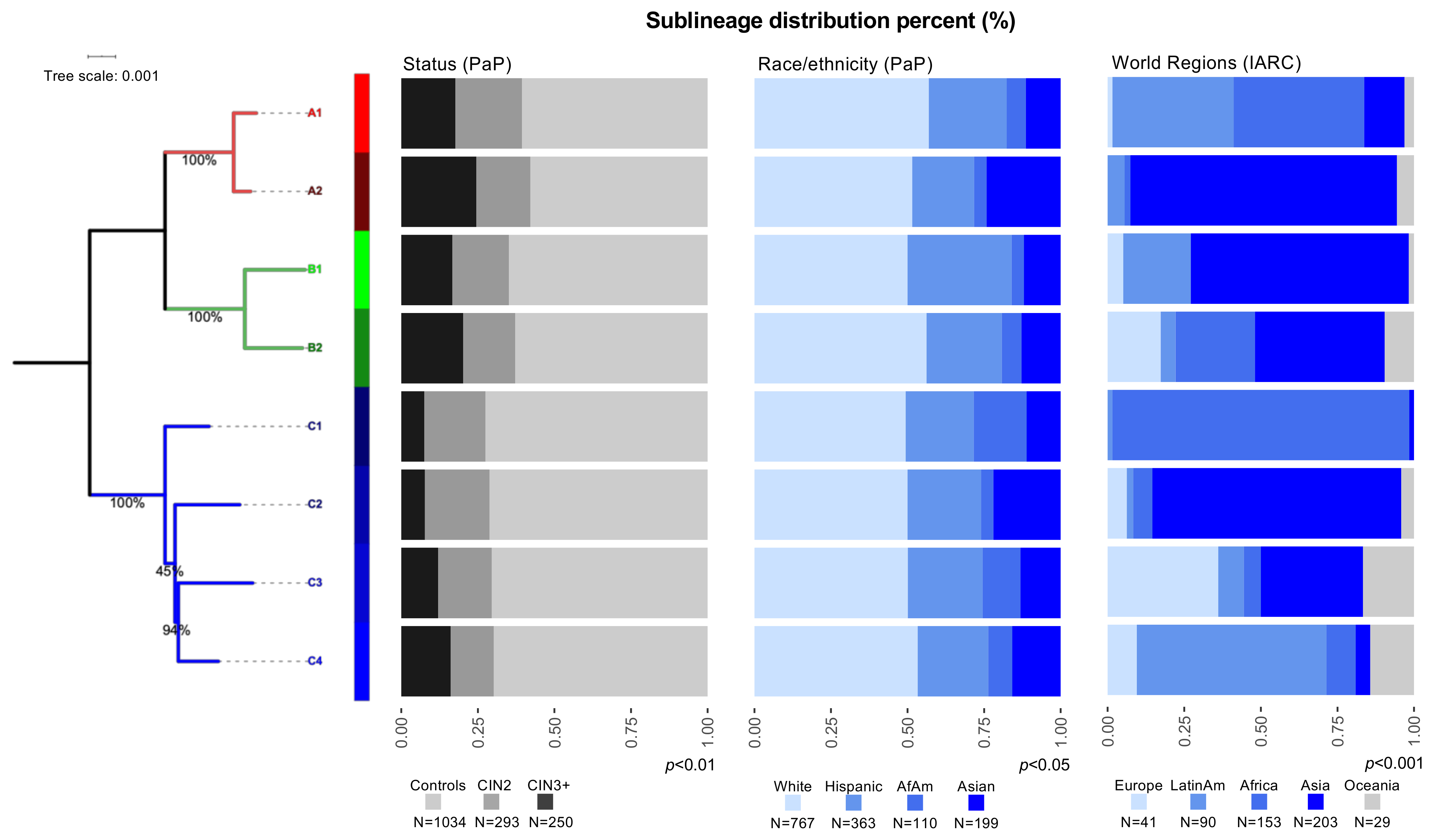
3.1. Distribution of HPV31 Lineages in PaP and IARC
3.2. HPV31 Lineages Are Associated with Precancer and Cancer
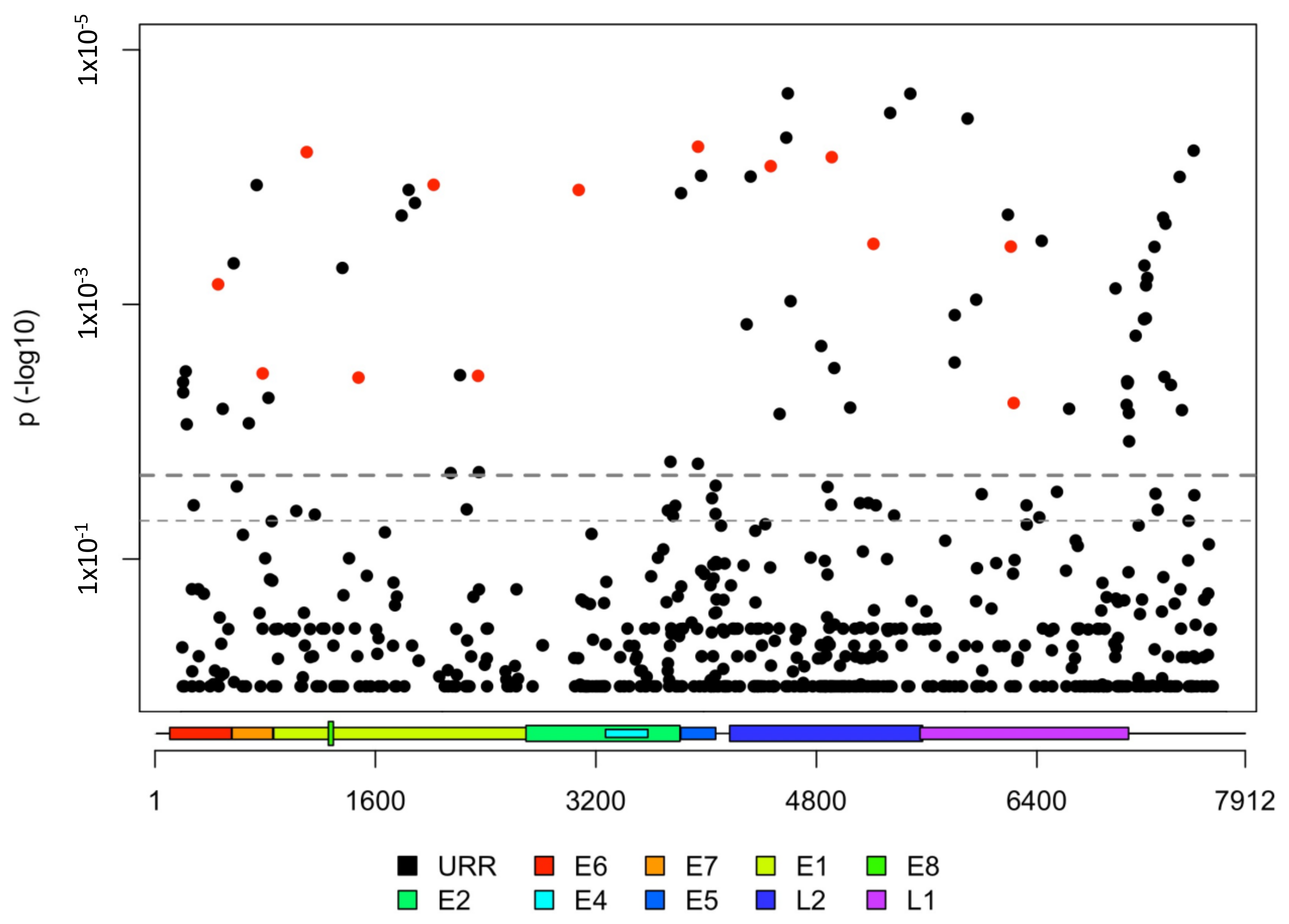
3.3. HPV31 Individual SNPs Are Associated with Cervical Carcinogenesis
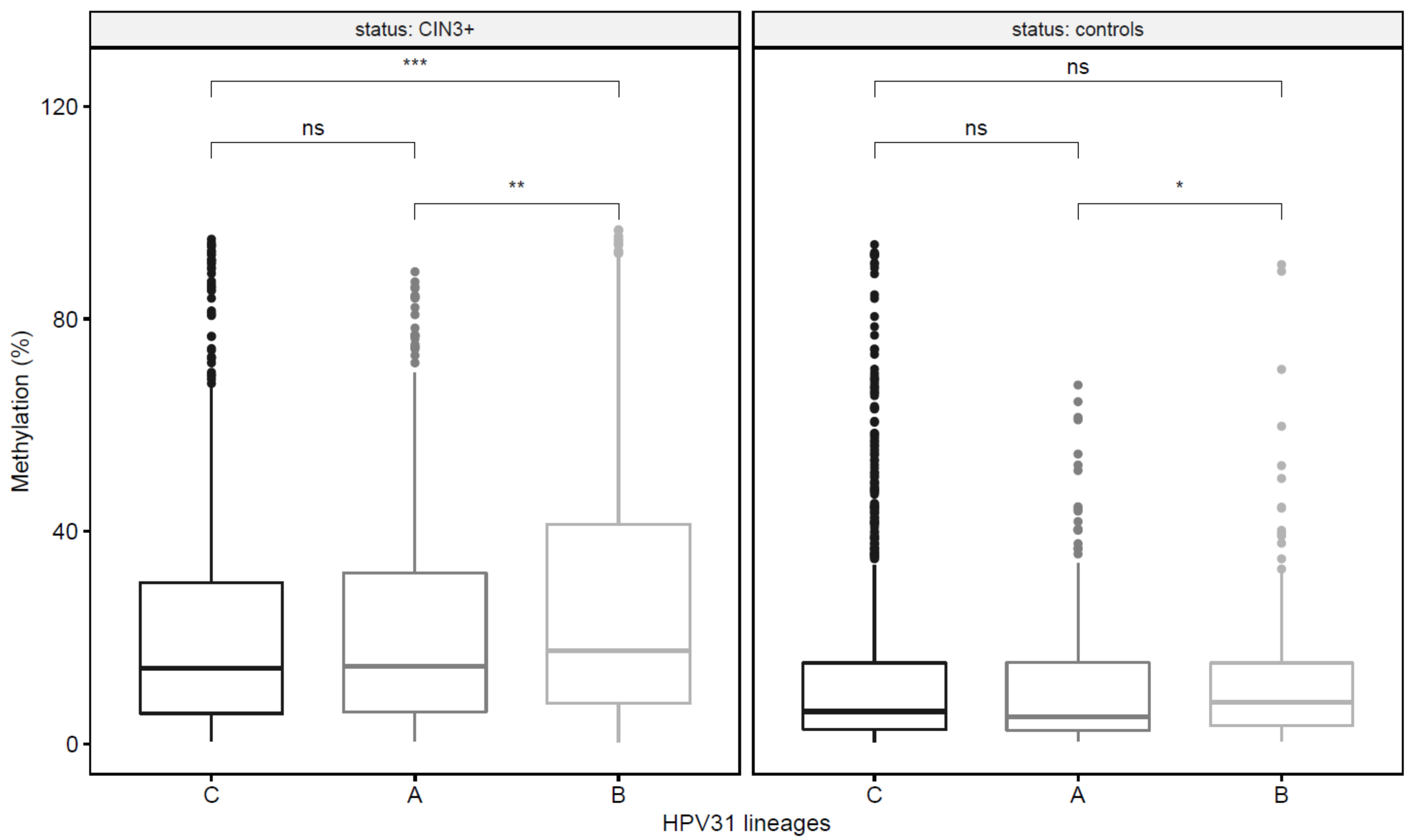
3.4. HPV31 Lineages Have Differing Methylation Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIS | adenocarcinoma in situ |
| AUC | areas under the curve |
| CI | confidence intervals |
| CIN | cervical intraepithelial neoplasia |
| CIN1 | cervical intraepithelial neoplasia grade 1 |
| CIN | cervical intraepithelial neoplasia grade 2 |
| CIN2+ | cervical intraepithelial neoplasia grade 2, grade 3 and cancer |
| CIN3 | cervical intraepithelial neoplasia grade 3 |
| CIN3+ | cervical intraepithelial neoplasia grade 3 and cancer |
| DNA | deoxyribonucleic acid |
| E1 | early gene 1 |
| E7 | early gene 7 |
| FDR | false discovery rate |
| FFPE | formalin fixed paraffin-embedded |
| HC2 | Hybrid Capture 2 |
| HPV | Human papillomavirus |
| HPV31 | Human papillomavirus type 31 |
| HR | high-risk |
| IARC | International Agency for Research on Cancer |
| ICC | invasive cervical cancer |
| KPNC | Kaiser Permanente Northern California |
| L1 | late gene 1 |
| MAF | minor allele frequency |
| MRCA | most recent common ancestor |
| NCI | National Cancer Institute |
| OR | odds ratio |
| PaP | Persistence and Progression |
| PCR | polymerase chain reaction |
| ROC | Receiver operating characteristic |
| SNPs | single nucleotide polymorphisms |
| STM | specimen transport medium |
| vs. | versus |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group. IARC Handbooks of Cancer Prevention: Cervix Cancer Screening; IARC Press: Lyon, France, 2005; Volume 10. [Google Scholar]
- WHO. Human papillomavirus vaccines: WHO position paper, October 2014. Wkly. Epidemiol. Rec. 2014, 89, 465–491. [Google Scholar]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [Green Version]
- PapillomaVirus Episteme (PaVe). Available online: https://pave.niaid.nih.gov/#home (accessed on 8 January 2021).
- De Sanjose, S.; Quint, W.G.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Schiffman, M.; Herrero, R.; Desalle, R.; Hildesheim, A.; Wacholder, S.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 2005, 337, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens--Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Goldsborough, M.D.; DiSilvestre, D.; Temple, G.F.; Lorincz, A.T. Nucleotide sequence of human papillomavirus type 31: A cervical neoplasia-associated virus. Virology 1989, 171, 306–311. [Google Scholar] [CrossRef]
- Arbyn, M.; Tommasino, M.; Depuydt, C.; Dillner, J. Are 20 human papillomavirus types causing cervical cancer? J. Pathol. 2014, 234, 431–435. [Google Scholar] [CrossRef]
- Smith, J.S.; Lindsay, L.; Hoots, B.; Keys, J.; Franceschi, S.; Winer, R.; Clifford, G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. Cancer 2007, 121, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Howell-Jones, R.; Li, N.; Bruni, L.; de Sanjose, S.; Franceschi, S.; Clifford, G.M. Human papillomavirus types in 115,789 HPV-positive women: A meta-analysis from cervical infection to cancer. Int. J. Cancer 2012, 131, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.C.; Schiffman, M.; Herrero, R.; Wacholder, S.; Hildesheim, A.; Castle, P.E.; Solomon, D.; Burk, R.; Proyecto Epidemiologico Guanacaste, G. Rapid clearance of human papillomavirus and implications for clinical focus on persistent infections. J. Natl. Cancer Inst. 2008, 100, 513–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molano, M.; Van den Brule, A.; Plummer, M.; Weiderpass, E.; Posso, H.; Arslan, A.; Meijer, C.J.; Munoz, N.; Franceschi, S.; The HPV Study Group. Determinants of clearance of human papillomavirus infections in Colombian women with normal cytology: A population-based, 5-year follow-up study. Am. J. Epidemiol. 2003, 158, 486–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, G.Y.; Bierman, R.; Beardsley, L.; Chang, C.J.; Burk, R.D. Natural history of cervicovaginal papillomavirus infection in young women. N. Engl. J. Med. 1998, 338, 423–428. [Google Scholar] [CrossRef]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Schiffman, M.; Wentzensen, N.; Wacholder, S.; Kinney, W.; Gage, J.C.; Castle, P.E. Human papillomavirus testing in the prevention of cervical cancer. J. Natl. Cancer Inst. 2011, 103, 368–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.A.; Gradissimo, A.; Schiffman, M.; Lam, J.; Sollecito, C.C.; Fetterman, B.; Lorey, T.; Poitras, N.; Raine-Bennett, T.R.; Castle, P.E.; et al. Human Papillomavirus DNA Methylation as a Biomarker for Cervical Precancer: Consistency across 12 Genotypes and Potential Impact on Management of HPV-Positive Women. Clin. Cancer Res. 2018, 24, 2194–2202. [Google Scholar] [CrossRef] [Green Version]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010, 70, 3159–3169. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations with Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J. Natl. Cancer Inst. 2016, 108, djw100. [Google Scholar] [CrossRef] [Green Version]
- Xi, L.F.; Schiffman, M.; Koutsky, L.A.; Hughes, J.P.; Hulbert, A.; Shen, Z.; Galloway, D.A.; Kiviat, N.B. Variant-specific persistence of infections with human papillomavirus Types 31, 33, 45, 56 and 58 and risk of cervical intraepithelial neoplasia. Int. J. Cancer 2016, 139, 1098–1105. [Google Scholar] [CrossRef] [Green Version]
- Xi, L.F.; Schiffman, M.; Koutsky, L.A.; Hughes, J.P.; Winer, R.L.; Mao, C.; Hulbert, A.; Lee, S.K.; Shen, Z.; Kiviat, N.B. Lineages of oncogenic human papillomavirus types other than type 16 and 18 and risk for cervical intraepithelial neoplasia. J. Natl. Cancer Inst. 2014, 106, dju270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Schiffman, M.; Herrero, R.; Desalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS ONE 2011, 6, e20183. [Google Scholar] [CrossRef]
- Mirabello, L.; Schiffman, M.; Ghosh, A.; Rodriguez, A.C.; Vasiljevic, N.; Wentzensen, N.; Herrero, R.; Hildesheim, A.; Wacholder, S.; Scibior-Bentkowska, D.; et al. Elevated methylation of HPV16 DNA is associated with the development of high grade cervical intraepithelial neoplasia. Int. J. Cancer 2013, 132, 1412–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantari, M.; Osann, K.; Calleja-Macias, I.E.; Kim, S.; Yan, B.; Jordan, S.; Chase, D.M.; Tewari, K.S.; Bernard, H.U. Methylation of human papillomavirus 16, 18, 31, and 45 L2 and L1 genes and the cellular DAPK gene: Considerations for use as biomarkers of the progression of cervical neoplasia. Virology 2014, 448, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Lorincz, A.T.; Brentnall, A.R.; Scibior-Bentkowska, D.; Reuter, C.; Banwait, R.; Cadman, L.; Austin, J.; Cuzick, J.; Vasiljevic, N. Validation of a DNA methylation HPV triage classifier in a screening sample. Int. J. Cancer 2016, 138, 2745–2751. [Google Scholar] [CrossRef] [Green Version]
- Louvanto, K.; Aro, K.; Nedjai, B.; Butzow, R.; Jakobsson, M.; Kalliala, I.; Dillner, J.; Nieminen, P.; Lorincz, A. Methylation in predicting progression of untreated high-grade cervical intraepithelial neoplasia. Clin. Infect. Dis 2019, 70, 2582–2590. [Google Scholar] [CrossRef]
- Wentzensen, N.; Sun, C.; Ghosh, A.; Kinney, W.; Mirabello, L.; Wacholder, S.; Shaber, R.; LaMere, B.; Clarke, M.; Lorincz, A.T.; et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. J. Natl. Cancer Inst. 2012, 104, 1738–1749. [Google Scholar] [CrossRef] [Green Version]
- Brentnall, A.R.; Vasiljevic, N.; Scibior-Bentkowska, D.; Cadman, L.; Austin, J.; Szarewski, A.; Cuzick, J.; Lorincz, A.T. A DNA methylation classifier of cervical precancer based on human papillomavirus and human genes. Int. J. Cancer 2014, 135, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Vasiljevic, N.; Scibior-Bentkowska, D.; Brentnall, A.; Cuzick, J.; Lorincz, A. A comparison of methylation levels in HPV18, HPV31 and HPV33 genomes reveals similar associations with cervical precancers. J. Clin. Virol. 2014, 59, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, P.E.; Shaber, R.; LaMere, B.J.; Kinney, W.; Fetterma, B.; Poitras, N.; Lorey, T.; Schiffman, M.; Dunne, A.; Ostolaza, J.M.; et al. Human papillomavirus (HPV) genotypes in women with cervical precancer and cancer at Kaiser Permanente Northern California. Cancer Epidemiol. Biomark. Prev. 2011, 20, 946–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaMere, B.J.; Howell, R.; Fetterman, B.; Shieh, J.; Castle, P.E.; Pap Cohort Study Group. Impact of 6-month frozen storage of cervical specimens in alkaline buffer conditions on human papillomavirus genotyping. J. Virol. Methods 2008, 151, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaMere, B.J.; Kornegay, J.; Fetterman, B.; Sadorra, M.; Shieh, J.; Castle, P.E.; PaP Cohort Study Group. Human papillomavirus genotyping after denaturation of specimens for Hybrid Capture 2 testing: Feasibility study for the HPV persistence and progression cohort. J. Virol. Methods 2007, 146, 80–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, F.X.; Manos, M.M.; Muñoz, N.; Sherman, M.; Jansen, A.M.; Peto, J.; Schiffman, M.H.; Moreno, V.; Kurman, R.; Shah, K.V. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. Natl. Cancer Inst. 1995, 87, 796–802. [Google Scholar] [CrossRef]
- Clifford, G.M.; Gallus, S.; Herrero, R.; Muñoz, N.; Snijders, P.J.; Vaccarella, S.; Anh, P.T.; Ferreccio, C.; Hieu, N.T.; Matos, E.; et al. Worldwide distribution of human papillomavirus types in cytologically normal women in the International Agency for Research on Cancer HPV prevalence surveys: A pooled analysis. Lancet 2005, 366, 991–998. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J.; The International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.V.; Walboomers, J.M.; Snijders, P.J.; Voorhorst, F.J.; Verheijen, R.H.; Fransen-Daalmeijer, N.; Meijer, C.J. Distribution of 37 mucosotropic HPV types in women with cytologically normal cervical smears: The age-related patterns for high-risk and low-risk types. Int. J. Cancer 2000, 87, 221–227. [Google Scholar] [CrossRef]
- Burk, R.D.; Ho, G.Y.; Beardsley, L.; Lempa, M.; Peters, M.; Bierman, R. Sexual behavior and partner characteristics are the predominant risk factors for genital human papillomavirus infection in young women. J. Infect. Dis. 1996, 174, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornet, I.; Gheit, T.; Franceschi, S.; Vignat, J.; Burk, R.D.; Sylla, B.S.; Tommasino, M.; Clifford, G.M.; the IARC HPV Variant Study Group. Human papillomavirus type 16 genetic variants: Phylogeny and classification based on E6 and LCR. J. Virol. 2012, 86, 6855–6861. [Google Scholar] [CrossRef] [Green Version]
- Cullen, M.; Boland, J.F.; Schiffman, M.; Zhang, X.; Wentzensen, N.; Yang, Q.; Chen, Z.; Yu, K.; Mitchell, J.; Roberson, D.; et al. Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res. 2015, 1, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Koster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Hamady, M.; Walker, J.J.; Harris, J.K.; Gold, N.J.; Knight, R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 2008, 5, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.W.; Hughes, A.L. Within-host nucleotide diversity of virus populations: Insights from next-generation sequencing. Infect. Genet. Evol. 2015, 30, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Jopling, C.; Boue, S.; Izpisua Belmonte, J.C. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjose, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ho, W.C.S.; Boon, S.S.; Law, P.T.Y.; Chan, M.C.W.; DeSalle, R.; Burk, R.D.; Chan, P.K.S. Ancient Evolution and Dispersion of Human Papillomavirus 58 Variants. J. Virol. 2017, 91, e01285-17. [Google Scholar] [CrossRef] [Green Version]
- Pimenoff, V.N.; de Oliveira, C.M.; Bravo, I.G. Transmission between Archaic and Modern Human Ancestors during the Evolution of the Oncogenic Human Papillomavirus 16. Mol. Biol. Evol. 2017, 34, 4–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; DeSalle, R.; Schiffman, M.; Herrero, R.; Wood, C.E.; Ruiz, J.C.; Clifford, G.M.; Chan, P.K.S.; Burk, R.D. Niche adaptation and viral transmission of human papillomaviruses from archaic hominins to modern humans. PLoS Pathog. 2018, 14, e1007352. [Google Scholar] [CrossRef]
- Wood, C.E.; Chen, Z.; Cline, J.M.; Miller, B.E.; Burk, R.D. Characterization and experimental transmission of an oncogenic papillomavirus in female macaques. J. Virol. 2007, 81, 6339–6345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeleine, M.M.; Johnson, L.G.; Smith, A.G.; Hansen, J.A.; Nisperos, B.B.; Li, S.; Zhao, L.P.; Daling, J.R.; Schwartz, S.M.; Galloway, D.A. Comprehensive analysis of HLA-A, HLA-B, HLA-C, HLA-DRB1, and HLA-DQB1 loci and squamous cell cervical cancer risk. Cancer Res. 2008, 68, 3532–3539. [Google Scholar] [CrossRef] [Green Version]
- Mirabello, L.; Frimer, M.; Harari, A.; McAndrew, T.; Smith, B.; Chen, Z.; Wentzensen, N.; Wacholder, S.; Castle, P.E.; Raine-Bennett, T.; et al. HPV16 methyl-haplotypes determined by a novel next-generation sequencing method are associated with cervical precancer. Int. J. Cancer 2015, 136, E146–E153. [Google Scholar] [CrossRef] [Green Version]
- Doeberitz, M.; Vinokurova, S. Host factors in HPV-related carcinogenesis: Cellular mechanisms controlling HPV infections. Arch. Med. Res. 2009, 40, 435–442. [Google Scholar] [CrossRef]
- Xi, L.F.; Koutsky, L.A.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Winer, R.L.; Ho, J.; Kiviat, N.B. Risk for high-grade cervical intraepithelial neoplasia associated with variants of human papillomavirus types 16 and 18. Cancer Epidemiol. Biomark. Prev. 2007, 16, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, M.; Gage, J.C.; Clifford, G.M.; Demarco, M.; Cheung, L.C.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, X.; et al. Association of HPV35 with cervical carcinogenesis among women of African ancestry: Evidence of viral-host interaction with implications for disease intervention. Int. J. Cancer 2020, 147, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.E.; Heimbrook, D.C.; Huber, H.E.; Wegrzyn, R.J.; Rotberg, N.S.; Stauffer, K.J.; Lumma, P.K.; Garsky, V.M.; Oliff, A. Specific N-methylations of HPV-16 E7 peptides alter binding to the retinoblastoma suppressor protein. J. Biol. Chem. 1992, 267, 908–912. [Google Scholar] [CrossRef]
- Armstrong, D.J.; Roman, A. The relative ability of human papillomavirus type 6 and human papillomavirus type 16 E7 proteins to transactivate E2F-responsive elements is promoter- and cell-dependent. Virology 1997, 239, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Schade, A.E.; Fischer, M.; DeCaprio, J.A. RB, p130 and p107 differentially repress G1/S and G2/M genes after p53 activation. Nucleic Acids Res. 2019, 47, 11197–11208. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, S.K.; van den Brule, A.J.; Paull, G.; Svare, E.I.; Sherman, M.E.; Thomsen, B.L.; Suntum, M.; Bock, J.E.; Poll, P.A.; Meijer, C.J. Type specific persistence of high risk human papillomavirus (HPV) as indicator of high grade cervical squamous intraepithelial lesions in young women: Population based prospective follow up study. BMJ 2002, 325, 572. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.R.; Shew, M.L.; Qadadri, B.; Neptune, N.; Vargas, M.; Tu, W.; Juliar, B.E.; Breen, T.E.; Fortenberry, J.D. A longitudinal study of genital human papillomavirus infection in a cohort of closely followed adolescent women. J. Infect. Dis. 2005, 191, 182–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [Green Version]
- Schiller, J.T.; Lowy, D.R. Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat. Rev. Microbiol. 2012, 10, 681–692. [Google Scholar] [CrossRef]
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L1. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Xiao, Y.; Yeager, M.; Clifford, G.; Wentzensen, N.; Cullen, M.; Boland, J.F.; Bass, S.; Steinberg, M.K.; Raine-Bennett, T.; et al. Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat. Commun. 2020, 11, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Lineages | Controls | CIN3+ | OR | 95% CI | p | |||
|---|---|---|---|---|---|---|---|---|
| N | % | N | % | |||||
| Lineage | ||||||||
| C | 521 | 50.4% | 89 | 35.6% | ref | |||
| A | 326 | 31.5% | 103 | 41.2% | 1.85 | 1.35 | 2.54 | |
| B | 187 | 18.1% | 58 | 23.2% | 1.82 | 1.25 | 2.63 | |
| Total | 1034 | 100.0% | 250 | 100.0% | ||||
| Sublineage | ||||||||
| C3 | 322 | 31.1% | 55 | 22.0% | ref | |||
| A1 | 267 | 25.8% | 78 | 31.2% | 1.71 | 1.17 | 2.5 | |
| A2 | 59 | 5.7% | 25 | 10.0% | 2.48 | 1.43 | 4.29 | |
| B1 | 35 | 3.4% | 9 | 3.6% | 1.51 | 0.69 | 3.31 | |
| B2 | 152 | 14.7% | 49 | 19.6% | 1.89 | 1.23 | 2.9 | |
| C1 | 58 | 5.6% | 6 | 2.4% | 0.61 | 0.25 | 1.47 | |
| C2 | 37 | 3.6% | 4 | 1.6% | 0.63 | 0.22 | 1.85 | |
| C4 | 104 | 10.1% | 24 | 9.6% | 1.35 | 0.8 | 2.29 | |
| Total | 1034 | 100.0% | 250 | 100.0% | ||||
| Race/Ethnicity versus all others | ||||||||
| A Lineage | ||||||||
| White | 143 | 50.4% | 64 | 63.4% | 1.71 | 1.07 | 2.72 | |
| Hispanic | 81 | 28.5% | 19 | 18.8% | 0.58 | 0.33 | 1.02 | |
| African-American | 18 | 6.3% | 3 | 3.0% | 0.45 | 0.13 | 1.57 | |
| Asian | 42 | 14.8% | 15 | 14.9% | 1.00 | 0.53 | 1.90 | 0.017 |
| Total | 284 | 100.0% | 101 | 100.0% | ||||
| B Lineage | ||||||||
| White | 90 | 54.9% | 29 | 53.7% | 0.95 | 0.51 | 1.77 | |
| Hispanic | 42 | 25.6% | 15 | 27.8% | 1.12 | 0.56 | 2.23 | |
| African-American | 11 | 6.7% | 3 | 5.6% | 0.82 | 0.22 | 3.05 | |
| Asian | 21 | 12.8% | 7 | 13.0% | 1.01 | 0.41 | 2.54 | 0.976 |
| Total | 164 | 100.0% | 54 | 100.0% | ||||
| C Lineage | ||||||||
| White | 237 | 49.0% | 48 | 57.1% | 1.39 | 0.87 | 2.22 | |
| Hispanic | 132 | 27.3% | 16 | 19.0% | 0.63 | 0.35 | 1.12 | |
| African-American | 51 | 10.5% | 6 | 7.1% | 0.65 | 0.27 | 1.57 | |
| Asian | 64 | 13.2% | 14 | 16.7% | 1.31 | 0.7 | 2.47 | 0.113 |
| Total | 484 | 100.0% | 84 | 100.0% | ||||
| Viral Gene/Region | No. Individuals with Variants (%) | p | p-FDR | |||
|---|---|---|---|---|---|---|
| All HPV31 lineages (n = 1284) | ||||||
| Controls (n = 1034) | CIN3+ cases (n = 250) | |||||
| E1 | 223 | 21.6% | 45 | 18.0% | 0.214 | 0.535 |
| E2 | 148 | 14.3% | 34 | 13.6% | 0.772 | 0.875 |
| E4 | 65 | 6.3% | 8 | 3.2% | 0.064 | 0.320 |
| E5 | 58 | 5.6% | 13 | 5.2% | 0.799 | 0.875 |
| E6 | 61 | 5.9% | 12 | 4.8% | 0.501 | 0.835 |
| E7 | 21 | 2.0% | 3 | 1.2% | 0.389 | 0.778 |
| L1 | 236 | 22.8% | 42 | 16.8% | 0.039 | 0.320 |
| L2 | 375 | 36.3% | 92 | 36.8% | 0.875 | 0.875 |
| URR | 181 | 17.5% | 41 | 16.4% | 0.679 | 0.875 |
| WG | 724 | 70.0% | 164 | 65.6% | 0.175 | 0.535 |
| HPV31 A lineages (n = 429) | ||||||
| Controls (n = 326) | CIN3+ cases (n = 103) | |||||
| E1 | 61 | 18.7% | 9 | 8.7% | 0.020 | 0.163 |
| E2 | 35 | 10.7% | 6 | 5.8% | 0.146 | 0.292 |
| E4 | 19 | 5.8% | 1 | 1.0% | 0.074 | 0.185 |
| E5 | 13 | 4.0% | 2 | 1.9% | 0.335 | 0.497 |
| E6 | 12 | 3.7% | 6 | 5.8% | 0.348 | 0.497 |
| E7 | 5 | 1.5% | 1 | 1.0% | 0.674 | 0.674 |
| L1 | 74 | 22.7% | 14 | 13.6% | 0.049 | 0.163 |
| L2 | 114 | 35.0% | 39 | 37.9% | 0.593 | 0.672 |
| URR | 53 | 16.3% | 19 | 18.4% | 0.605 | 0.672 |
| WG | 225 | 69.0% | 60 | 58.3% | 0.045 | 0.163 |
| Gene | Lineage | CpG Site † | Controls (n = 85) | CIN3+ (n = 89) | Difference | p * | p-FDR | AUC | 95% CI | OR | 95% CI | p # | p-FDR | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Median | n | Median | ||||||||||||||
| E2 | A | 3414 | 22 | 1.89 | 32 | 6.19 | 4.30 ** | 6.6 × 10−7 | 4.4 × 10−5 | 0.90 | 0.82 | 0.98 | 22.62 | 5.16 | 99.19 | 3.5 × 10−5 | 0.001 |
| B | 3414 | 13 | 5.47 | 28 | 7.77 | 2.30 | 0.560 | 0.560 | 0.56 | 0.37 | 0.74 | 1.81 | 0.47 | 6.97 | 0.390 | 0.410 | |
| C | 3414 | 50 | 2.67 | 29 | 5.29 | 2.62 | 0.035 | 0.049 | 0.64 | 0.52 | 0.77 | 3.38 | 1.29 | 8.81 | 0.013 | 0.020 | |
| L2 | A | 5530 | 22 | 3.97 | 32 | 11.88 | 7.92 | 0.005 | 0.011 | 0.73 | 0.58 | 0.88 | 9.52 | 2.71 | 33.51 | 4.5 × 10−4 | 0.003 |
| B | 5530 | 9 | 4.33 | 27 | 19.41 | 15.08 ** | 0.002 | 0.007 | 0.84 | 0.66 | 1.00 | 25.00 | 3.39 | 184.50 | 0.002 | 0.004 | |
| C | 5530 | 45 | 4.32 | 23 | 11.56 | 7.24 | 0.004 | 0.009 | 0.72 | 0.59 | 0.85 | 5.14 | 1.69 | 15.63 | 0.004 | 0.007 | |
| L2 | A | 5521 | 22 | 2.66 | 32 | 5.40 | 2.74 | 0.004 | 0.009 | 0.73 | 0.58 | 0.88 | 8.00 | 2.33 | 27.46 | 9.5 × 10−4 | 0.004 |
| B | 5521 | 9 | 3.65 | 27 | 6.62 | 2.97 | 0.096 | 0.110 | 0.69 | 0.46 | 0.91 | 5.50 | 1.07 | 28.20 | 0.041 | 0.056 | |
| C | 5521 | 45 | 3.10 | 23 | 6.26 | 3.16 ** | 6.0 × 10−5 | 0.002 | 0.80 | 0.69 | 0.91 | 7.20 | 2.24 | 23.17 | 9.3 × 10−4 | 0.004 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinheiro, M.; Harari, A.; Schiffman, M.; Clifford, G.M.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Raine-Bennett, T.; Steinberg, M.; et al. Phylogenomic Analysis of Human Papillomavirus Type 31 and Cervical Carcinogenesis: A Study of 2093 Viral Genomes. Viruses 2021, 13, 1948. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101948
Pinheiro M, Harari A, Schiffman M, Clifford GM, Chen Z, Yeager M, Cullen M, Boland JF, Raine-Bennett T, Steinberg M, et al. Phylogenomic Analysis of Human Papillomavirus Type 31 and Cervical Carcinogenesis: A Study of 2093 Viral Genomes. Viruses. 2021; 13(10):1948. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101948
Chicago/Turabian StylePinheiro, Maisa, Ariana Harari, Mark Schiffman, Gary M. Clifford, Zigui Chen, Meredith Yeager, Michael Cullen, Joseph F. Boland, Tina Raine-Bennett, Mia Steinberg, and et al. 2021. "Phylogenomic Analysis of Human Papillomavirus Type 31 and Cervical Carcinogenesis: A Study of 2093 Viral Genomes" Viruses 13, no. 10: 1948. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101948