
Genetic Characterizations and Molecular Evolution of the Measles Virus Genotype B3’s Hemagglutinin (H) Gene in the Elimination Era


Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Genetic and Amino Acid Diversity of MeV Genotype B3’s H Gene
2.3. Time-Evolutionary Pattern of MeV Genotype B3’s H Gene
2.4. Time-Scale Phylogenetic and Phylodynamic Analysis
3. Results
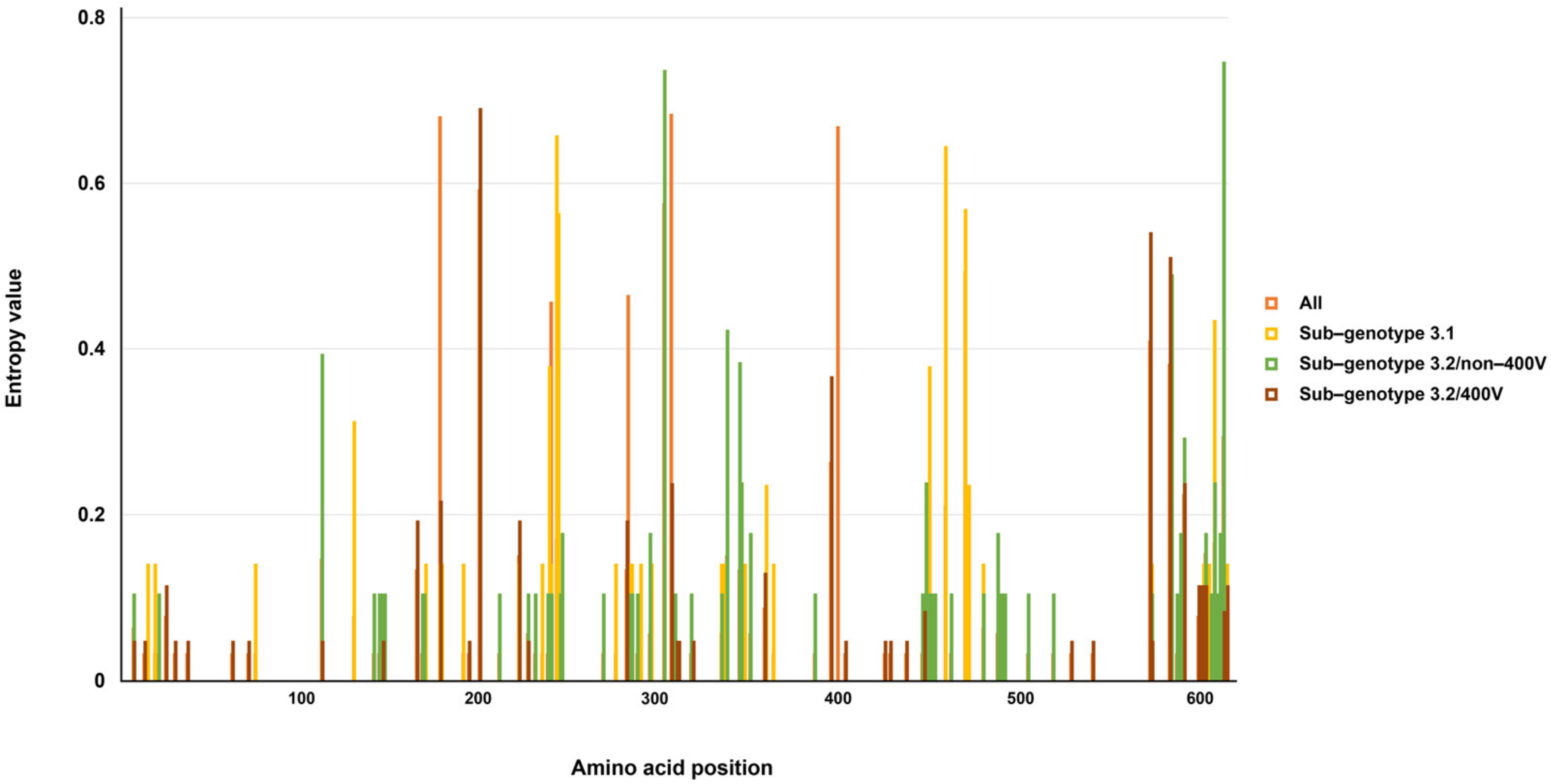
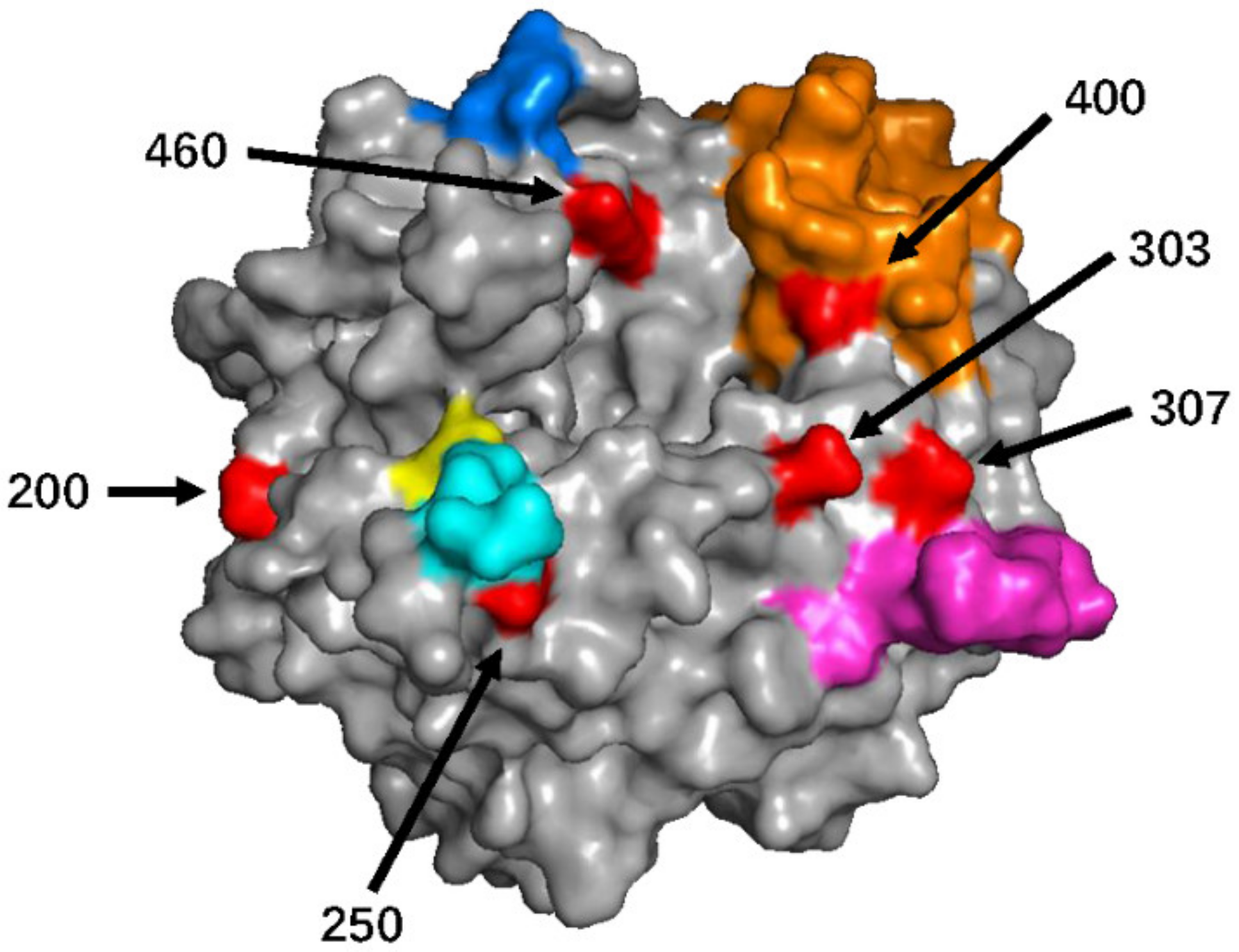
3.1. Genetic and Amino Acid Variation of MeV Genotype B3’s H Gene
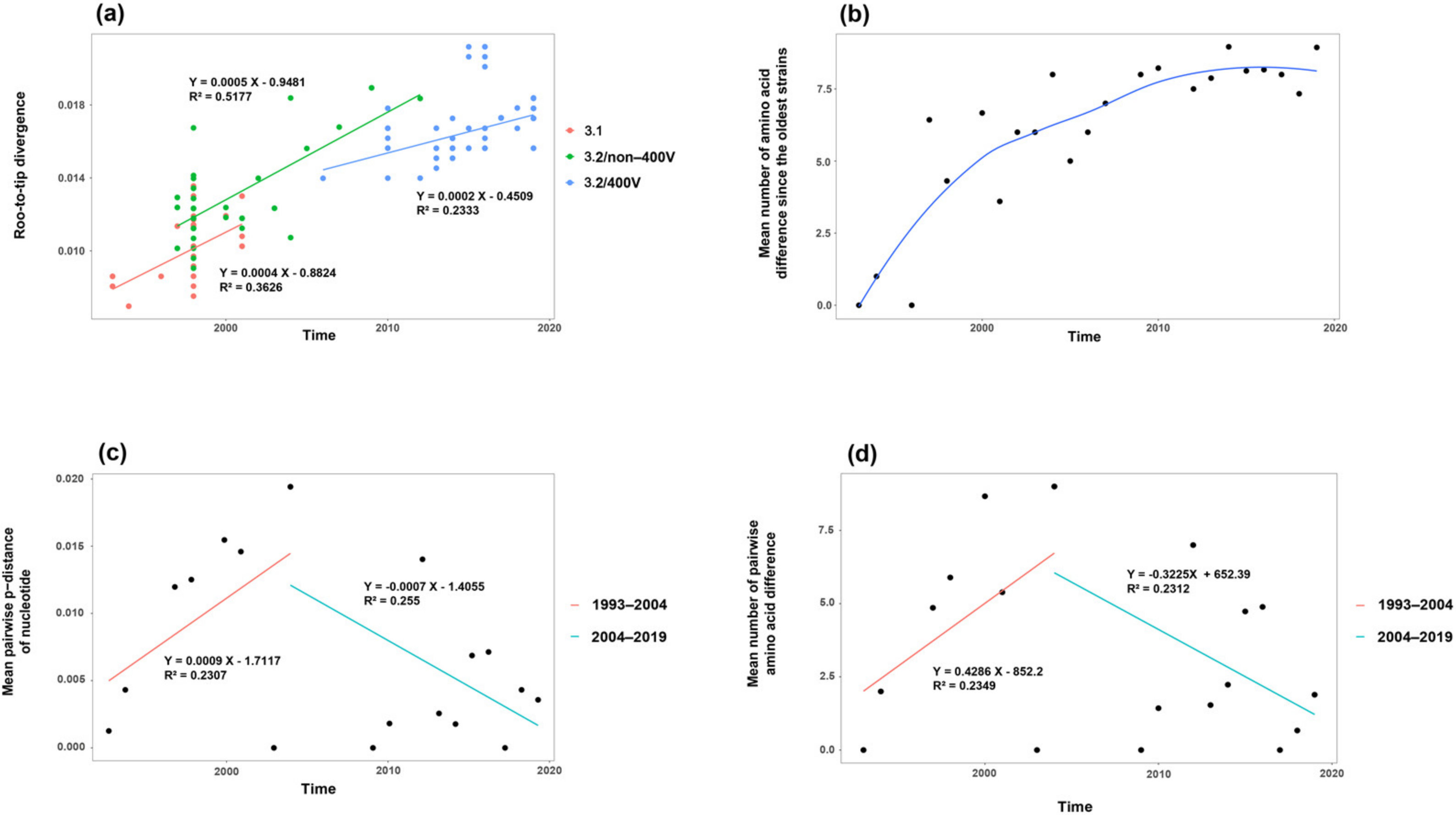
3.2. Time-Evolutionary Pattern of MeV Genotype B3’s H Gene
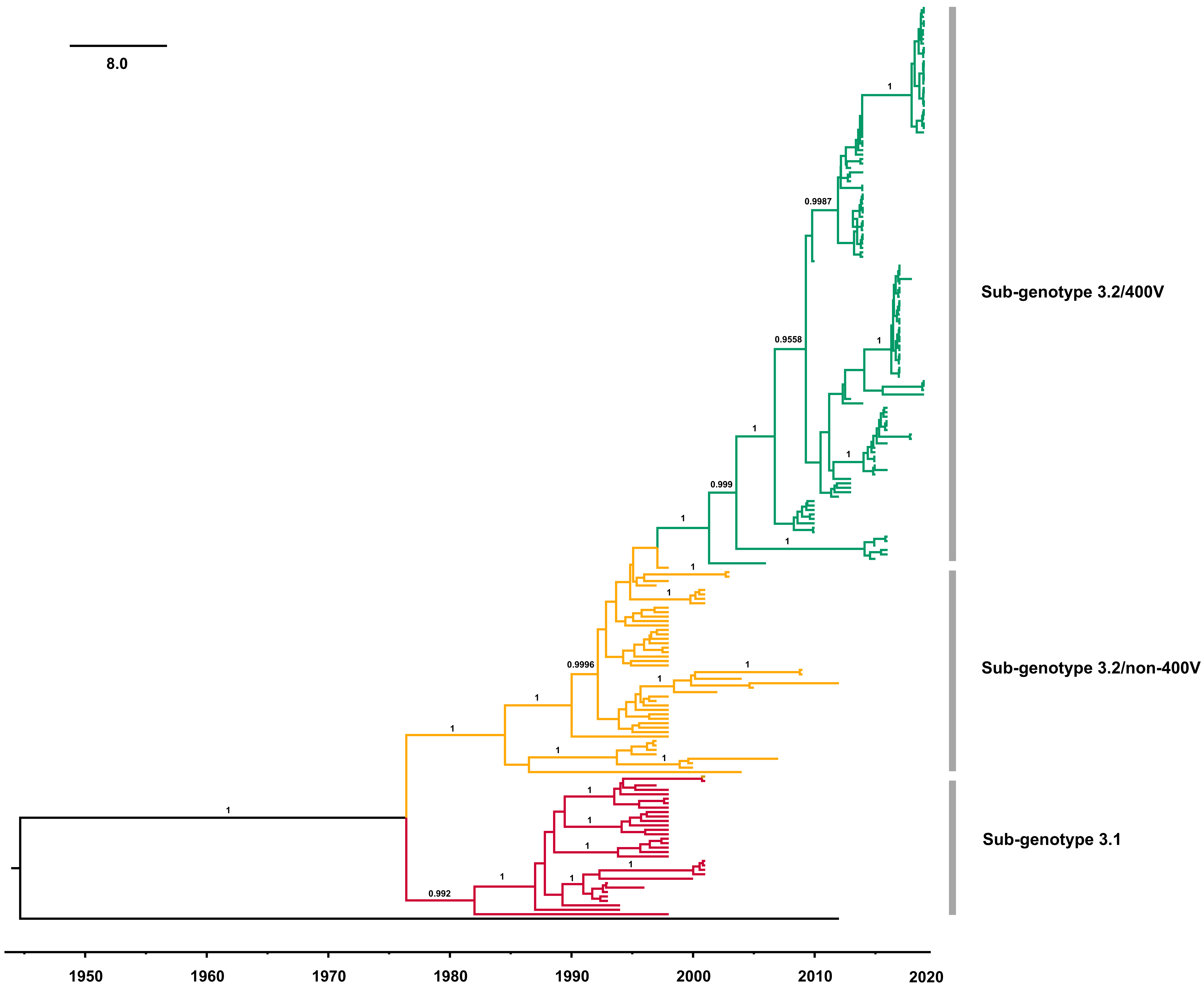
3.3. Time-Scale Phylogenetic and Phylodynamic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moss, W.J. Measles. Lancet 2017, 390, 2490–2502. [Google Scholar] [CrossRef]
- Dabbagh, A.; Laws, R.L.; Steulet, C.; Dumolard, L.; Mulders, M.N.; Kretsinger, K.; Alexander, J.P.; Rota, P.A.; Goodson, J.L. Progress toward regional measles elimination—Worldwide, 2000–2017. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1323–1329. [Google Scholar] [CrossRef]
- Patel, M.K.; Dumolard, L.; Nedelec, Y.; Sodha, S.V.; Steulet, C.; Gacic-Dobo, M.; Kretsinger, K.; McFarland, J.; Rota, P.A.; Goodson, J.L. Progress toward regional measles elimination—Worldwide, 2000–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 1105–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.K.; Goodson, J.L.; Alexander, J.P., Jr.; Kretsinger, K.; Sodha, S.V.; Steulet, C.; Gacic-Dobo, M.; Rota, P.A.; McFarland, J.; Menning, L.; et al. Progress toward regional measles elimination—Worldwide, 2000–2019. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1700–1705. [Google Scholar] [CrossRef]
- Komabayashi, K.; Seto, J.; Tanaka, S.; Suzuki, Y.; Ikeda, T.; Onuki, N.; Yamada, K.; Ahiko, T.; Ishikawa, H.; Mizuta, K. The largest measles outbreak, including 38 modified measles and 22 typical measles cases in its elimination era in Yamagata, Japan, 2017. Jpn. J. Infect. Dis. 2018, 71, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Ayodele, O.; Abbasi, A.F.; Prakash, S.; Gosse, J.; Younis, S.; Mangat, J.; Chan, H. Measles outbreak in unvaccinated and partially vaccinated children and adults in the United States and Canada (2018–2019): A narrative review of cases. Inquiry 2019, 56, 46958019894098. [Google Scholar] [CrossRef]
- Zhao, S.; Han, L.; He, D.; Qin, J. Public awareness, news promptness and the measles outbreak in Hong Kong from March to April, 2019. Infect. Dis. 2020, 52, 284–290. [Google Scholar] [CrossRef]
- Roberts, L. How COVID hurt the fight against other dangerous diseases. Nature 2021, 592, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Méndez, E.; Arias, C. Astroviruses. In Fields Virology, 6th ed.; Knipe, D., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- World Health Organization. Measles virus nomenclature update: 2012. Wkly. Epidemiol. Rec. 2012, 87, 73–81. [Google Scholar]
- Brown, K.E.; Rota, P.A.; Goodson, J.L.; Williams, D.; Abernathy, E.; Takeda, M.; Mulders, M.N. Genetic characterization of measles and rubella viruses detected through global measles and rubella elimination surveillance, 2016–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Currie, J.; Davies, L.; McCarthy, J.; Perry, M.; Moore, C.; Cottrell, S.; Bowley, M.; Williams, C.; Shankar, A.G.; Stiff, R. Measles outbreak linked to european B3 outbreaks, Wales, United Kingdom, 2017. Eurosurveillance 2017, 22, 17-00673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogka, V.; Horefti, E.; Evangelidou, M.; Kostaki, E.G.; Paraskevis, D.; Flountzi, A.; Georgakopoulou, T.; Magaziotou, I.; Mentis, A.; Karamitros, T. Spatiotemporal distribution and genetic characterization of measles strains circulating in Greece during the 2017–2018 outbreak. Viruses 2020, 12, 1166. [Google Scholar] [CrossRef]
- Zaidi, S.S.Z.; Hameed, A.; Suleman Rana, M.; Alam, M.M.; Umair, M.; Aamir, U.B.; Hussain, M.; Sharif, S.; Shaukat, S.; Angez, M.; et al. Identification of measles virus genotype B3 associated with outbreaks in Islamabad, Pakistan, 2013-2015. J. Infect. Public Health 2018, 11, 540–545. [Google Scholar] [CrossRef]
- Ackley, S.F.; Hacker, J.K.; Enanoria, W.T.A.; Worden, L.; Blumberg, S.; Porco, T.C.; Zipprich, J. Genotype-specific measles transmissibility: A branching process analysis. Clin. Infect. Dis. 2018, 66, 1270–1275. [Google Scholar] [CrossRef] [Green Version]
- De Swart, R.L.; Yuksel, S.; Osterhaus, A.D. Relative contributions of measles virus hemagglutinin- and fusion protein-specific serum antibodies to virus neutralization. J. Virol. 2005, 79, 11547–11551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. The role of extended and whole genome sequencing for tracking transmission of measles and rubella viruses: Report from the Global Measles and Rubella Laboratory Network meeting, 2017. Wkly. Epidemiol. Rec. 2018, 93, 55–59. [Google Scholar]
- Kimura, H.; Saitoh, M.; Kobayashi, M.; Ishii, H.; Saraya, T.; Kurai, D.; Tsukagoshi, H.; Shirabe, K.; Nishina, A.; Kozawa, K.; et al. Molecular evolution of haemagglutinin (H) gene in measles virus. Sci. Rep. 2015, 5, 11648. [Google Scholar] [CrossRef]
- Bianchi, S.; Canuti, M.; Ciceri, G.; Gori, M.; Colzani, D.; Dura, M.; Pennati, B.M.; Baggieri, M.; Magurano, F.; Tanzi, E.; et al. Molecular epidemiology of B3 and D8 measles viruses through hemagglutinin phylogenetic history. Int. J. Mol. Sci. 2020, 21, 4435. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit_a user-friendly biological sequence alignment editor and analysis program for Windows 95_98_NT. Nucleic Acids Res. Symp Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Maino, T.; Jean-Philippe, B.; Kazuhiko, K.; Katsumi, M.; Claude, M.; Makoto, T. Measles virus hemagglutinin protein epitopes: The basis of antigenic stability. Viruses 2016, 8, 216. [Google Scholar]
- World Health Organization. Nomenclature for describing the genetic characteristics of wild-type measles viruses (update). Wkly. Epidemiol. Rec. 2001, 76, 242–247. [Google Scholar]
- Hanses, F.; Truong, A.T.; Ammerlaan, W.; Ikusika, O.; Adu, F.; Oyefolu, A.O.; Omilabu, S.A.; Muller, C.P. Molecular epidemiology of Nigerian and Ghanaian measles virus isolates reveals a genotype circulating widely in western and central Africa. J. Gen. Virol. 1999, 80, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Kouomou, D.W.; Nerrienet, E.; Mfoupouendoun, J.; Tene, G.; Whittle, H.; Wild, T.F. Measles virus strains circulating in Central and West Africa: Geographical distribution of two B3 genotypes. J. Med. Virol. 2002, 68, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Seki, F.; Miyoshi, M.; Ikeda, T.; Nishijima, H.; Saikusa, M.; Itamochi, M.; Minagawa, H.; Kurata, T.; Ootomo, R.; Kajiwara, J.; et al. Nationwide molecular epidemiology of measles virus in Japan between 2008 and 2017. Front. Microbiol. 2019, 10, 1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.S.Z.; Hameed, A.; Ali, N.; Umair, M.; Alam, M.M.; Rana, M.S.; Sharif, S.; Aamir, U.B.; Shaukat, S.; Angez, M.; et al. A measles outbreak in Sindh, Pakistan caused by a genotype B3 virus. Arch. Virol. 2017, 162, 3603–3610. [Google Scholar] [CrossRef] [PubMed]
- Fatemi Nasab, G.S.; Salimi, V.; Abbasi, S.; Adjami Nezhad Fard, F.; Mokhtari Azad, T. Comparison of neutralizing antibody titers against outbreak-associated measles genotypes (D4, H1 and B3) in Iran. Pathog. Dis. 2016, 74, ftw089. [Google Scholar] [CrossRef] [PubMed]
- Bartz, R.; Brinckmann, U.; Dunster, L.M.; Rima, B.; Ter Meulen, V.; Schneider-Schaulies, J. Mapping amino acids of the measles virus hemagglutinin responsible for receptor (CD46) downregulation. Virology 1996, 224, 334–337. [Google Scholar] [CrossRef]
- Hashiguchi, T.; Ose, T.; Kubota, M.; Maita, N.; Kamishikiryo, J.; Maenaka, K.; Yanagi, Y. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat. Struct. Mol. Biol. 2011, 18, 135–141. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, G.; Qi, J.; Li, Y.; He, Y.; Xu, X.; Shi, J.; Zhang, C.W.; Yan, J.; Gao, G.F. Structure of measles virus hemagglutinin bound to its epithelial receptor nectin-4. Nat. Struct. Mol. Biol. 2013, 20, 67–72. [Google Scholar] [CrossRef]
- Hu, A.; Cattaneo, R.; Schwartz, S.; Norrby, E. Role of N-linked oligosaccharide chains in the processing and antigenicity of measles virus haemagglutinin protein. J. Gen. Virol. 1994, 75, 1043–1052. [Google Scholar] [CrossRef]
- Hashiguchi, T.; Kajikawa, M.; Maita, N.; Takeda, M.; Kuroki, K.; Sasaki, K.; Kohda, D.; Yanagi, Y.; Maenaka, K. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. USA 2007, 104, 19535–19540. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.Y.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-dependent rates of molecular evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.R.; Brown, K.E.; Jin, L.; Santibanez, S.; Shulga, S.V.; Aboudy, Y.; Demchyshyna, I.V.; Djemileva, S.; Echevarria, J.E.; Featherstone, D.F.; et al. High genetic diversity of measles virus, World Health Organization European Region, 2005-2006. Emerg. Infect. Dis. 2008, 14, 107–114. [Google Scholar] [CrossRef]
- Ji, Y.; Xu, S.; Zhang, Y.; Zhu, Z.; Mao, N.; Jiang, X.; Ma, C.; Lu, P.; Wang, C.; Liang, Y.; et al. Genetic characterization of wild-type measles viruses isolated in China, 2006–2007. Virol. J. 2010, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Rima, B.K.; Earle, J.A.; Baczko, K.; ter Meulen, V.; Liebert, U.G.; Carstens, C.; Carabaña, J.; Caballero, M.; Celma, M.L.; Fernandez-Muñoz, R. Sequence divergence of measles virus haemagglutinin during natural evolution and adaptation to cell culture. J. Gen. Virol. 1997, 78, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Takeda, M.; Gotoh, K.; Takeuchi, F.; Sekizuka, T.; Kuroda, M.; Mizuta, K.; Ryo, A.; Tanaka, R.; Ishii, H.; et al. Molecular evolution of hemagglutinin (H) gene in measles virus genotypes D3, D5, D9, and H1. PLoS ONE 2012, 7, e50660. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, Y.; Zhu, Z.; Liu, C.; Mao, N.; Ji, Y.; Wang, H.; Jiang, X.; Li, C.; Tang, W.; et al. Genetic characterization of the hemagglutinin genes of wild-type measles virus circulating in china, 1993–2009. PLoS ONE 2013, 8, e73374. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Alía, M.Á.; Fernández-Muñoz, R.; Casasnovas, J.M.; Porras-Mansilla, R.; Serrano-Pardo, Á.; Pagán, I.; Ordobás, M.; Ramírez, R.; Celma, M.L. Measles virus genetic evolution throughout an imported epidemic outbreak in a highly vaccinated population. Virus Res. 2015, 196, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Faneye, A.O.; Motayo, B.O.; Adeniji, J.A. Molecular characterization and evolutionary dynamics of measles virus sequences isolated from children in Lagos and Ibadan, South Western, Nigeria. J. Infect. Public Health 2020, 13, 309–312. [Google Scholar] [CrossRef]
- Ciceri, G.; Canuti, M.; Bianchi, S.; Gori, M.; Piralla, A.; Colzani, D.; Libretti, M.; Frati, E.R.; Baggieri, M.; Lai, A.; et al. Genetic variability of the measles virus hemagglutinin gene in B3 genotype strains circulating in Northern Italy. Infect. Genet. Evol. 2019, 75, 103943. [Google Scholar] [CrossRef]
- Charlesworth, B. Fundamental concepts in genetics: Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Durrheim, D.N.; Andrus, J.K.; Tabassum, S.; Bashour, H.; Githanga, D.; Pfaff, G. A dangerous measles future looms beyond the COVID-19 pandemic. Nat. Med. 2021, 27, 360–361. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Frequency in All Analyzed B3 Strains | Frequency in Specific Cluster |
|---|---|---|
| 471 E | 84.88% (174/205) | Sub-genotype 3.2: 99.43% (173/174) |
| 240 N | 84.39% (173/205) | Sub-genotype 3.2: 99.42% (172/173) |
| 283 G | 83.90% (172/205) | Sub-genotype 3.2: 100% (172/172) |
| 303 D | 76.10% (156/205) | Sub-genotype 3.2: 100% (156/156) |
| 400 V | 61.46% (126/205) | Sub-genotype 3.2/400V: 100% (126/126) |
| 178 T | 58.54% (120/205) | Sub-genotype 3.2/400V: 94.44% (119/120) |
| 307 I | 57.56% (118/205) | Sub-genotype 3.2/400V: 100% (118/118) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, N.; Li, M.; Huang, Y.; Zhou, L.; Wang, B. Genetic Characterizations and Molecular Evolution of the Measles Virus Genotype B3’s Hemagglutinin (H) Gene in the Elimination Era. Viruses 2021, 13, 1970. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101970
Zhou N, Li M, Huang Y, Zhou L, Wang B. Genetic Characterizations and Molecular Evolution of the Measles Virus Genotype B3’s Hemagglutinin (H) Gene in the Elimination Era. Viruses. 2021; 13(10):1970. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101970
Chicago/Turabian StyleZhou, Nan, Mingma Li, Yue Huang, Lu Zhou, and Bei Wang. 2021. "Genetic Characterizations and Molecular Evolution of the Measles Virus Genotype B3’s Hemagglutinin (H) Gene in the Elimination Era" Viruses 13, no. 10: 1970. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101970