
Genomic Signatures in HPV-Associated Tumors


1
Department of Radiation Oncology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
2
Department of Radiation Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA 02189, USA
*
Author to whom correspondence should be addressed.
Viruses 2021, 13(10), 1998; https://0-doi-org.brum.beds.ac.uk/10.3390/v13101998
Submission received: 17 September 2021
/
Revised: 30 September 2021
/
Accepted: 2 October 2021
/
Published: 5 October 2021
(This article belongs to the Special Issue Viral Infection: A Threat for Genomic Stability in Host Cells)
Abstract
:Papillomaviruses dysregulate the G1/S cell cycle transition in order to promote DNA synthesis in S phase, which is a requirement for viral replication. The human papillomaviruses (HPV) E6 and E7 oncoproteins mediate degradation of the cell cycle regulators p53 and Rb, which are two of the most universally disrupted tumor-suppressor genes in all of cancer. The G1/S checkpoint is activated in normal cells to allow sufficient time for DNA repair in G1 before proceeding to replicate DNA and risk propagating unrepaired errors. The TP53 pathway suppresses a variety of such errors, including translocation, copy number alterations, and aneuploidy, which are thus found in HPV-associated tumors similarly to HPV-negative tumors with other mechanisms of TP53 disruption. However, E6 and E7 maintain a variety of other virus–host interactions that directly disrupt a growing list of other DNA repair and chromatin remodeling factors, implying HPV-specific repair deficiencies. In addition, HPV-associated squamous cell carcinomas tumors clinically respond differently to DNA damaging agents compared to their HPV negative counterparts. The focus of this review is to integrate three categories of observations: (1) pre-clinical understanding as to the effect of HPV on DNA repair, (2) genomic signatures of DNA repair in HPV-associated tumor genomes, and (3) clinical responses of HPV-associated tumors to DNA damaging agents. The goals are to try to explain why HPV-associated tumors respond so well to DNA damaging agents, identify missing pieces, and suggest clinical strategies could be used to further improve treatment of these cancers.
1. Introduction
The high-risk human papillomaviruses (HR-HPV) infect the basal layer of epithelium in the anogenital region and the oropharynx. While most infections are cleared, in some individuals, HR-HPV infections and the introduction of oncoproteins E6 and E7 is the founding carcinogenic event for the development of squamous cell carcinomas arising from these anatomic locations. While sufficient for the immortalization of keratinocytes, E6 and E7 are insufficient for transformation into malignancy, and the acquisition of additional driver alterations is required. In murine models, the expression of HPV18 E6/E7 alone does not induce malignancy, which also requires an additional oncogene (ras or fos) [1]. In human HPV-associated cervical, oropharyngeal, and anal cancers, these additional drivers include copy number alterations and driver gene mutations affecting tumor suppressor genes or oncogenes [2,3,4]. In addition, numerous passenger alterations in non-driver elements are also acquired. Inferences about how these driver and passenger alterations are formed can be gleaned by evaluating patterns or “signatures” of mutations and structural rearrangements [5,6]. A signature is a reflection of an underlying genomic insult (e.g., a pyrimidine dimer from UV light) and how the early cancer cell repairs that insult. Since the E6/E7 oncoprotein expression is a first step in carcinogenesis, the impact of these oncoproteins on fundamental processes such as DNA repair and genomic stability can be discerned through measurement of the genomic signatures acquired in cancer genomes on the course from HPV infection to clinical tumor detection. Two of the most important features of the oncoproteins E6 and E7 include the inactivation of p53 and Rb; however, virtually all cancers also have acquired mechanisms of inactivation of these targets as well. For instance, nearly all HPV− head and neck squamous cell carcinomas have acquired TP53 mutations and loss of CDKN2A [2], providing an alternative means of bypassing the Rb-mediated restriction point and the G1/S checkpoint enforced by p53. Thus, evaluating genomic differences between HPV+ and HPV− cancer genomes can help distinguish between E6/E7 effects on DNA repair and genomic stability other than simply inactivation/loss of TP53. These comparisons between HPV+ vs. HPV− tumor genomes comparison can best be done between head and neck squamous cell carcinomas as the overwhelming majority of cervical cancers and most anal cancers are HPV+.
2. Radiosensitivity Differences between HPV+ and HPV− Tumors
Locoregionally advanced head and neck squamous cell carcinomas are often best managed with concurrent chemoradiation, consisting of 70 Gy in 2 Gy fractions with concurrent cisplatin either weekly or in three bolus doses over the course of radiotherapy [7,8]. The value of the cisplatin in addition to radiotherapy was demonstrated in multiple randomized trials, [7,8] including two recent dedicated HPV-associated oropharyngeal SCC trials [9,10]. Concurrent cisplatin is similarly supported by phase III trials in squamous cell carcinomas of the cervix [11,12,13], and in the case of anal canal squamous cell carcinoma, the crosslinking agent mitomycin C substituted for a platinum salt also improves outcomes [14,15]. However, the primary curative modality is the radiotherapy since chemotherapy alone is non-curative, but the addition of concurrent cisplatin improves the 10-year locoregional cure rate in HNSCC from 54 to 71% compared to radiation alone [8].
In 2008, a landmark analysis of a prospective Eastern Cooperative Oncology Group (ECOG) trial of concurrent chemoradiation for head and neck cancers established superior outcomes for HPV-associated squamous cell carcinomas in terms of local control following radiotherapy as well as overall survival [16]. The superior radiosensitivity of HPV-associated HNSCC compared to HPV negative HNSCC has been confirmed in multiple other trials [17,18] and is also observed in SCC of the cervix [19,20], vulva [21,22,23], and anal canal [24,25,26], suggesting a consistent biology. Using HPV association as a biomarker, multiple phase II trials have now supported radiation dose reductions to 60 Gy [27,28], 54 Gy [29,30], or even 30 Gy [31], which is not possible in HPV-negative HNSCC. However, recent studies have shown that patients with integrated HPV genomes (a subset of 20% of all HPV-positive patients) have poor clinical outcomes compared to patients with episomal HPV; thus, we might need to further stratify patients before selecting them for de-escalation therapy [32,33].
Pre-clinical models of HPV-associated HNSCC are few, but multiple groups have replicated the demonstrated increased radiation sensitivity in collections of HPV+ HNSCC cell lines compared to HPV− HNSCC cell lines using gold standard clonogenic survival assays [34,35]. In patient-derived xenograft models, HPV+ HSNCC similarly are generally more radiosensitive compared to HPV− models [36]. These comparisons are made between collections of genetically heterogeneous cell lines, but isogenic comparisons of cell lines with and without E6 and E7 expressed also demonstrated HPV oncogene-mediated radiation sensitivity [37]. To compare radiation responses in implanted xenografts, Brezar et al. constructed an isogenic pair of the commonly used HPV negative HNSCC cell line FaDu and an FaDu line expressing E6/E7 [38]. Following 10 Gy of radiation, the E6/E7 FaDu xenograft exhibited a five-fold growth delay relative to the parental FaDu xenograft [38].
3. Proposed Mechanisms for Increased Radiosensitivity in HPV+ Tumors
The cell-intrinsic radiosensitivity found in clonogenic survival assays has been linked to a DNA double-strand break repair defect. Again using a comparison of cell line collections in HPV+ and HPV− categories, the resolution of γH2AX foci after radiation is delayed in HPV+ cell lines, suggesting a defect in DSBR (double strand break repair) [34,35]. Isogenic comparisons also confirm a defect, and when E6/E7 or E7 alone are expressed in immortalized keratinocytes or transgenetically introduced into skin keratinocytes in a murine model, the resolution of γH2AX is delayed [37]. Finally, when E6/E7 are introduced into a primary model of oral cancer, γH2AX is increased [39].
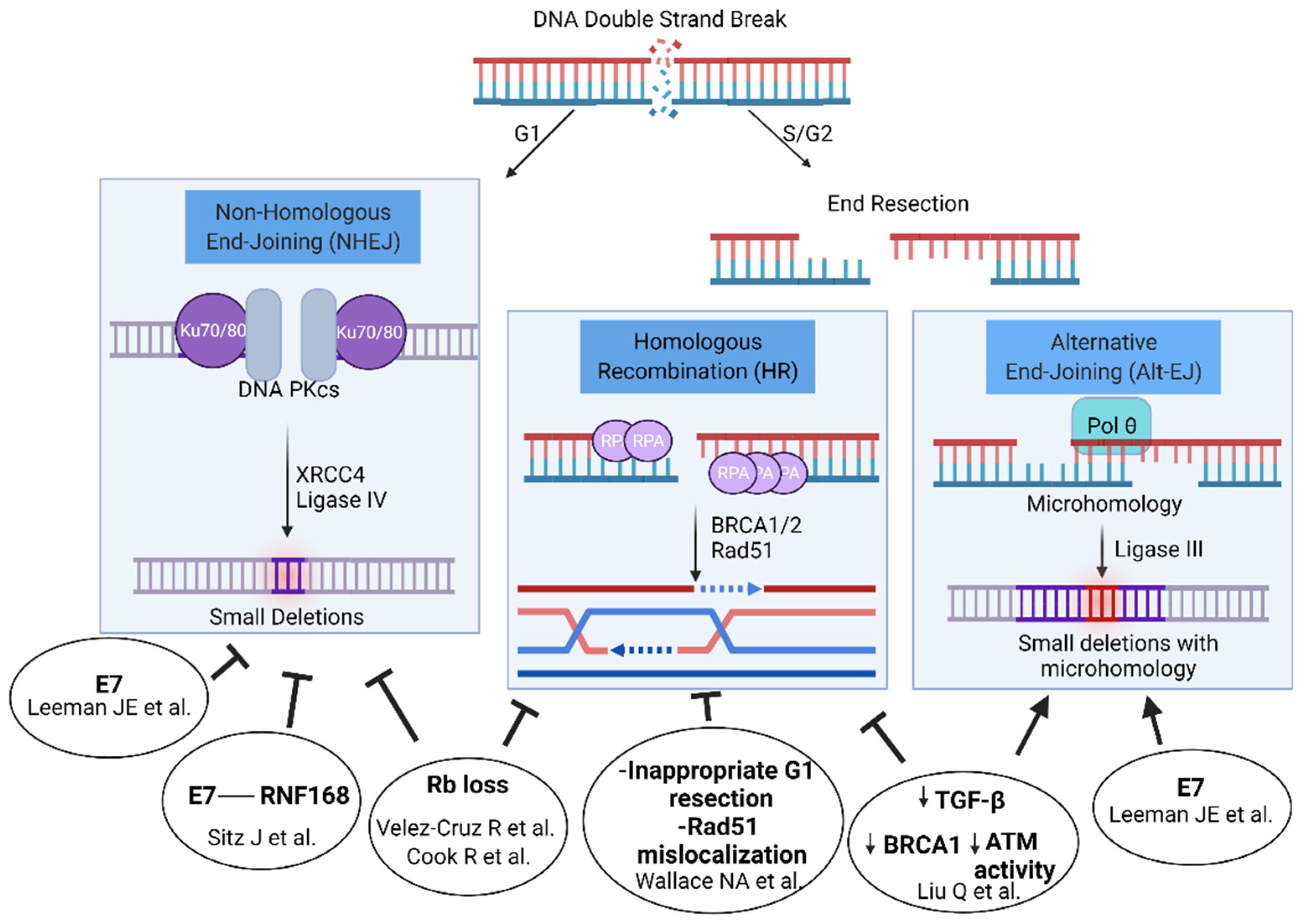
Both E6 or E7 individually and joint E6/E7 expression have been linked to DSBR defects and multiple DSBR pathways are proposed to be abrogated, including both homologous recombination [40,41] and nonhomologous end-joining [42,43]. Thus, there may not be one single explanatory mechanism but rather multiple (see Figure 1). Both E6 and E7 increased persistent γH2AX signal after radiation when expressed individually in the HPV-negative FaDu cell line [44]. However, primarily E7 delayed resolution of γH2AX in immortalized keratinocytes and transgenic models of E6 and E7 expression in skin [37] and only E7 or E6/E7 led to a DSB defect in an isogenic U2OS cell line model [42].
Following a double-strand break created by irradiation, an estimated 70% of breaks are repaired through nonhomologous end joining, a rapidly completed pathway consisting of end recognition by the XRCC5/6 heterodimer and DNA-PK, which forms a synaptic complex across the break site. Compatible ends can be directly ligated through Ligase IV, or limited end processing can occur to remove oxidatively damaged residues before subsequent ligation. In S and G2 cells, CTIP and the MRN complex can initiate end-resection, leaving a single strand 3′ prime end, which can invade a sister chromatid strand, find homology, and complete homologous recombination. A less well-characterized pathway, alternative end joining (Alt-EJ), also makes use of the exposed single stranded 3′ end from each side of the break. Small stretches of homology between opposite strands of the break, termed ‘microhomology’, can be annealed, initiating repair through polymerization and the clipping of non-complementary tails.
HPV E6/E7 expression has been linked to homologous recombination deficiency. Liu et al. have described an abrogation of TGF-β signaling in HPV+ HNSCCs and a subsequent de-repression of miR-182, which in turn inhibits BRCA1 and FOXO3 expression, thereby inhibiting homologous recombination directly through BRCA1 and indirectly through FOXO3 effects on ATM activity [41]. Inhibition of TGF-β signaling suppressed homologous recombination, increasing alternative end joining. Furthermore, when the expression of TGF-β genes are analyzed pan-cancer, this TGF-β signature is inversely correlated with expression of alt-EJ genes. An impact on ATM activation was also seen through E6-mediated degradation of TIP60, which is an acetyltransferase involved in the activation of ATM [45]. E7 also activates STAT5, causing increased phosphorylation of both ATM and ATR [46,47]. Furthermore, this activation of the JAK/STAT pathway helps HPV evade the immune system, and thus, targeting the JAK/STAT pathway could be another strategy to treat HPV-associated tumors [48]. Wallace et al. similarly demonstrated the inhibition of homologous recombination when E6 is expressed in a U2OS cell background, a 50% reduction in homologous recombination through an inappropriate initiation of homologous recombination in G1, and mislocalization of Rad51 away from DNA damage foci [40]. Additionally, the E7 oncoprotein induces the overexpression of the p16ink4a (p16) cell cycle inhibitor protein, and p16 may play indirect roles in homologous recombination [49]. p16 inhibits CCND1 expression, which in addition to its role in cell cycle progression also is proposed to have a direct role in homologous recombination [50]. p16 overexpression also decreases TRIP12 expression, an E3 ubiquitin ligase, and subsequently the overaccumulation of RNF168 [51]. Finally, the Rb protein itself (which is degraded by E7) has been demonstrated to promote HR through recruitment of the BRG1 ATPase, increasing end resection possibly through reducing the density of nucleosomes surrounding a DSB [52]. Thus, a number of different proposed mechanisms link E6/E7 oncoproteins to an HR defect.
In contrast, when comparing panels of HPV+ and HPV− cell lines that did exhibit separable radiation sensitivities in the hands of multiple groups [34,35], no such difference can be observed in cisplatin sensitivity [53]. Cisplatin mediates cytotoxicity through interstrand crosslinks, which require an active HR pathway to resolve. Response to platinum is a hallmark feature of HR-deficient cancers such as germline mutated BRCA1/2 breast and ovarian cancers.
The E6/E7 oncoproteins have also been proposed to influence repair through end joining. Using three different methods, our group measured the impact of E7 on DSBR and found an E7-mediated suppression of NHEJ, with a concomitant increase in Alt-EJ and HR, similar to the phenotype of NHEJ-inhibiting drugs [42,54]. The mechanism may be explained by the effect of E7 on viral replication centers, which are extrachromosomal DNA structures formed during the life cycle of the virus. A number of DNA repair proteins are recruited to viral replication centers, including ATM, the MRN complex, BRCA1, and 53BP1 [55,56,57,58]. Sitz et al. found that E7 binds RNF168, and the interaction is required for viral replication [43]. RNF168 ubiquitinates the histone protein H2A [59], which is one of two important histone markers that recruit 53BP1 to double-strand breaks. The 53BP1/RIF1/shieldin complex axis serves as the primary mechanism to protect the ends of a double-strand break from resection, thus promoting nonhomologous end joining over homologous recombination [60,61,62,63]. E7 expression substantially reduced 53BP1 foci in response to radiation (a marker of NHEJ) and promoted homologous recombination through a reporter cassette assay [43]. Another possible mechanism lies in the Rb protein itself. Rb is also reported to mediate the recruitment of XRCC5/6, key NHEJ effectors, to double-strand breaks, and Rb knockdown reduces NHEJ efficiency by cassette reporters [64].
While many studies have focused on how E6 and E7 modulate DSBR, viral replication itself requires DDR activation [56], including activation of ATM and ATR signaling pathways [46,57,65,66]. Homologous recombination proteins are also required for productive replication [65], and various DSBR factors are recruited to viral replicating centers, including ATM, BRCA1, RAD51, NBS1, ATM, WRN, and TOPBP1 [67,68,69,70]. The E1 and E2 proteins induce the DNA damage response as seen by increased γH2AX foci and DSBs measured by COMET assay in S and G2 phases [67,71]. In addition, the E2 protein can bind directly to several proteins involved in the DNA damage response and modulate their function, including TopBP1 [68,72]. These observations imply that viral replication may involve DNA structure complexity that requires DNA repair to proceed. A plurality of HPV-associated cancers maintains episomal DNA copies of HPV genomes, which could be replication-competent [2,3,32,33], and E2, E4, and E5 can remain expressed in some HPV+ cancers [73]. How replication competence affects overall DSBR in HPV+ tumors is unclear, and a worthwhile comparison would be episomal only vs. integrated HPV+ cancers in terms of sensitivity to DNA damaging agents and DSBR pathway utilization.
In addition to the DNA repair related, cell-intrinsic radiosensitivity mechanisms, other possibilities include incomplete TP53 inactivation [34], expression of radiosensitizing E6 isoforms [74], or prolonged G2/M arrest [35]; there are also cell-extrinsic proposed mechanisms involving the tumor microenvironment, such as decreased tumor hypoxia and improved immunoreactivity, which are nicely reviewed elsewhere [39,75,76].
4. Genomics Signatures in HPV Cancers
A complementary approach to cell-based analyses of DSBR is to analyze cancer genomes to measure characteristics DNA repair scars (“signatures”) that can reflect the underlying DNA repair capacity of the tumor. Examples include cancers defective in mismatch repair exhibiting microsatellite instability in the genome and cancers defective in homologous recombination exhibiting a variety of associated genomic alterations. What can the underlying genomics of HPV+ cancers teach us about DNA repair in these cancers?
First, we know that the expression of E6/E7 into cells can cause genomic instability, which is reflected by rearrangements, translocations, and amplifications [77,78], and also changes in ploidy [79]. Of course, all these measures of genomic and chromosomal instability are associated with TP53 loss, which is also mediated through E6. HPV− tumors exhibit near universal TP53 genetic inactivation. When HPV+ and HPV− HNSCC genomes are directly compared, HPV+ HNSCC actually exhibit fewer copy number alterations compared HPV− HNSCC genomes (median 113 vs. 136, p = 0.026) and a higher percentage of HPV+ HNSCC genomes are classified as “M” type, driven by more mutations in cancer drivers than copy number changes (58% vs. 27%) [2]. Thus, it is not clear from raw measures that HPV+ genomes exhibit genomic instability over and above that of other tumors, such as HPV− tumors with TP53 loss (see Table 1).
Second, the overall frequency of mutations does not differ between HPV+ and HPV− HNSCC genomes. However, the mode of acquisition of single base substitutions does differ substantially. HPV− genomes are dominated by C>A substitutions induced by smoking, whereas HPV+ genomes exhibit dominance of the APOBEC signature involving C>T and C>G mutations in TpCpN trinucleotides. The APOBEC cytidine deaminases mediate anti-viral activity through deamination of T to U in single-stranded viral DNA. As a harmful by-product, single-stranded DNA occurring in the genome during replication can similarly be affected by APOBEC enzymes, inducing base substitutions.
Third, when focusing on genomic signatures present in cancer deficient in homologous recombination, HPV+ tumors do not exhibit SBS3, the single base substitution pattern found in BRCA1/2 biallelic mutated cancers defective in HR [80], and if anything, the SBS3 contribution is slightly higher in HPV-negative HNSCCs [42,80,81]. Furthermore, BRCA1/2 mutated cancers exhibit an increase in large-scale state transition (LST) score, which is reflective of large rearrangements [82], but HPV+ HNSCC genomes do not exhibit this hallmark, either [42].
What is different between HPV+ and HPV− tumors is the frequency of deletions associated with microhomology-based repair used in alternative end joining [42]. Those HPV+ tumors with the highest levels of E7 expression exhibited a still higher percentage of Alt-EJ like scars. Finally, HPV+ tumors with high levels with ALT-EJ genomic scars exhibited improved disease-free survival following radiation (3-year DFS 60.1 vs. 41.2%, p = 0.04) [42]. Subsequently a prevalence of Alt-EJ genomic scars (termed ID6 in this case) without SBS3 was similarly demonstrated in cervical cancer whole genomes [83]. To reconcile the absence of HR-associated base substitution (SBS3) and large-scale state transition (LST) signatures and the presence of Alt-EJ deletion signatures, one explanation could be that the Alt-EJ signatures are reflective of defective NHEJ. Alternative end joining was first discovered in V(D)J junctions within lymphocytes deficient in NHEJ [84,85] and thus is a backup to canonical NHEJ, just as it is a backup to homologous recombination. Finally, the Alt-EJ signature and its relation to clinical outcomes in HPV+ HNSCCs was further confirmed in a prospective study [86].
5. Clinical Responses to Platinum in HPV+ Cancer
Another means of assessing real-world homologous recombination deficiency is to compare the clinical response rates of recurrent and metastatic HPV+ tumors to platinum salts and PARP inhibitors to those of HPV− tumors. Platinum and PARP inhibitor sensitivity are largely determined by HR capacity. For instance, the overall response rates to platinum in germline bi-allelic mutant BRCA1/2 cancers range from 65 to 95% [87,88,89,90,91,92]. In germline BRCA1/2 pancreas cancers, the ORR to cisplatin/5-FU is 65% [87,88]. In prostate cancer, these BRCA1/2 cases respond to carboplatin/taxol at a rate of 75% [89]. In breast cancer, not typically treated with platinum salts, 68% of biallelic mutant BRCA1/2 cases respond to cisplatin/gemcitabine [90]. Finally, in ovarian cancer, germline BRCA1/2 cases respond in 87–96% of cases in prospective trials [91,92]. In HPV+ HNSCCs, the clinical response rate was recently measured in the standard of care cisplatin/5-FU arm of the practice changing EXTREME trial. For all HNSCCs, the ORR in HPV+ cases was only 22%, which is marginally different that the 17% response rate observed in HPV− cases. When narrowed to only the oropharyngeal HPV+ cases, the ORR was 24% compared to 21% in HPV− disease [93]. Thus, the response rates of HPV+ tumors to platinum salts are not similar to true HR-deficient cancers, although a smaller, relative HR defect cannot be excluded.
6. Summary and Discussions
The radiosensitivity of HPV+ HNSCC, anal and vulvar cancers is a clinically important topic, as few other biomarkers of radiation sensitivity exist. In pre-clinical models, there is a considerable body of work supporting both HR and NHEJ deficiency induced by E6/E7. HPV+ tumor genomes demonstrate an absence of canonical HR deficiency signatures, such as SBS3 and LST, but they do exhibit increased Alt-EJ scars relative to their HPV− counterparts, possibly supporting increased Alt-EJ in response to a partial NHEJ deficiency. Finally, available clinical data strongly support a role for cisplatin in concurrent chemoradiotherapy, but as a stand-alone agent, it does not result in the kind of response rates common in true HR-deficient cancers. Opportunities for improvement in this field include additional prospective validation of Alt-EJ signatures and clinical investigation of DNA damage response inhibitors that can best take advantage of HPV-associated DSBR defects.
7. Patents
JEL and DSH are listed inventors on a patent filed by Memorial Sloan Kettering Cancer Center regarding the use of alternative end-joining genomic signatures to predict radiation sensitivity in connection with prior work [42]. There are no licenses or royalties.
Author Contributions
Conceptualization, D.S.H., J.E.L., S.S.H., D.L.; writing—original draft preparation, D.S.H.; writing—review and editing, D.S.H., J.E.L., S.S.H., D.L.; visualization, D.L.; supervision, D.S.H.; funding acquisition, D.S.H. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in the paper was supported by research grants from the National Cancer Institute [R33 CA236670-01A1], Emerson Collective Cancer Research Fund, the American Association for Cancer Research AstraZeneca START fellowship [19-40-12-HUSS] and in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute [P30CA008748]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
J.E.L. and D.S.H. are listed inventors on a patent filed by Memorial Sloan Kettering Cancer Center regarding the use of alternative end-joining genomic signatures to predict radiation sensitivity. There are no licenses or royalties. DSH has a research contract with SQZBiotechnologies for an unrelated project and has received travel funds from Biorad, Inc.
References
- Pei, X.F.; Meck, J.M.; Greenhalgh, D.; Schlegel, R. Cotransfection of HPV-18 and v-fos DNA induces tumorigenicity of primary human keratinocytes. Virology 1993, 196, 855–860. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helen, F.; Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Chung, J.H.; Sanford, E.; Johnson, A.; Klempner, S.J.; Schrock, A.B.; Palma, N.A.; Erlich, R.L.; Frampton, G.M.; Chalmers, Z.R.; Vergilio, J.; et al. Comprehensive genomic profiling of anal squamous cell carcinoma reveals distinct genomically defined classes. Ann. Oncol. 2016, 27, 1336–1341. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef] [PubMed]
- Setton, J.; Reis-Filho, J.S.; Powell, S.N. Homologous recombination deficiency: How genomic signatures are generated. Curr. Opin. Genet. Dev. 2021, 66, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Adelstein, D.J.; Li, Y.; Adams, G.L.; Wagner, H., Jr.; Kish, J.A.; Ensley, J.F.; Schuller, D.E.; Forastiere, A.A. An intergroup phase III comparison of standard radiation therapy and two schedules of concurrent chemoradiotherapy in patients with unresectable squamous cell head and neck cancer. J. Clin. Oncol. 2003, 21, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forastiere, A.A.; Zhang, Q.; Weber, R.S.; Maor, M.H.; Goepfert, H.; Pajak, T.F.; Morrison, W.; Glisson, B.; Trotti, A.; Ridge, J.A.; et al. Long-term results of RTOG 91-11: A comparison of three nonsurgical treatment strategies to preserve the larynx in patients with locally advanced larynx cancer. J. Clin. Oncol. 2013, 31, 845–852. [Google Scholar] [CrossRef]
- Gillison, M.L.; Trotti, A.M.; Harris, J.; Eisbruch, A.; Harari, P.M.; Adelstein, D.J.; Jordan, R.C.K.; Zhao, W.; Sturgis, E.M.; Burtness, B.; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet 2019, 393, 40–50. [Google Scholar] [CrossRef]
- Mehanna, H.; Robinson, M.; Hartley, A.; Kong, A.; Foran, B.; Fulton-Lieuw, T.; Dalby, M.; Mistry, P.; Sen, M.; O’Toole, L.; et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): An open-label randomised controlled phase 3 trial. Lancet 2019, 393, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef]
- Morris, M.; Eifel, P.J.; Lu, J.; Grigsby, P.W.; Levenback, C.; Stevens, R.E.; Rotman, M.; Gershenson, D.M.; Mutch, D.G. Pelvic radiation with concurrent chemotherapy compared with pelvic and para-aortic radiation for high-risk cervical cancer. N. Engl. J. Med. 1999, 340, 1137–1143. [Google Scholar] [CrossRef]
- Green, J.A.; Kirwan, J.M.; Tierney, J.F.; Symonds, P.; Fresco, L.; Collingwood, M.; Williams, C.J. Survival and recurrence after concomitant chemotherapy and radiotherapy for cancer of the uterine cervix: A systematic review and meta-analysis. Lancet 2001, 358, 781–786. [Google Scholar] [CrossRef]
- Bartelink, H.; Roelofsen, F.; Eschwege, F.; Rougier, P.; Bosset, J.F.; Gonzalez, D.G.; Peiffert, D.; van Glabbeke, M.; Pierart, M. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: Results of a phase III randomized trial of the European Organization for Research and Treatment of Cancer Radiotherapy and Gastrointestinal Cooperative Groups. J. Clin. Oncol. 1997, 15, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Northover, J.; Glynne-Jones, R.; Sebag-Montefiore, D.; James, R.; Meadows, H.; Wan, S.; Jitlal, M.; Ledermann, J. Chemoradiation for the treatment of epidermoid anal cancer: 13-year follow-up of the first randomised UKCCCR Anal Cancer Trial (ACT I). Br. J. Cancer 2010, 102, 1123–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhry, C.; Westra, W.H.; Li, S.; Cmelak, A.; Ridge, J.A.; Pinto, H.; Forastiere, A.; Gillison, M.L. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl. Cancer Inst. 2008, 100, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Rischin, D.; Young, R.J.; Fisher, R.; Fox, S.B.; Le, Q.T.; Peters, L.J.; Solomon, B.; Choi, J.; O’Sullivan, B.; Kenny, L.M.; et al. Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J. Clin. Oncol. 2010, 28, 4142–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Carunchio, L.; Soveral, I.; Steenbergen, R.D.; Torne, A.; Martinez, S.; Fuste, P.; Pahisa, J.; Marimon, L.; Ordi, J.; del Pino, M. HPV-negative carcinoma of the uterine cervix: A distinct type of cervical cancer with poor prognosis. BJOG 2015, 122, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Wakeham, K.; Kavanagh, K.; Cuschieri, K.; Millan, D.; Pollock, K.G.; Bell, S.; Burton, K.; Reed, N.S.; Graham, S.V. HPV status and favourable outcome in vulvar squamous cancer. Int. J. Cancer 2017, 140, 1134–1146. [Google Scholar] [CrossRef]
- Hay, C.M.; Lachance, J.A.; Lucas, F.L.; Smith, K.A.; Jones, M.A. Biomarkers p16, Human Papillomavirus and p53 Predict Recurrence and Survival in Early Stage Squamous Cell Carcinoma of the Vulva. J. Low Genit. Tract. Dis. 2016, 20, 252–256. [Google Scholar] [CrossRef]
- Sand, F.L.; Nielsen, D.M.B.; Frederiksen, M.H.; Rasmussen, C.L.; Kjaer, S.K. The prognostic value of p16 and p53 expression for survival after vulvar cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2018. [Google Scholar] [CrossRef]
- Horne, Z.D.; Dohopolski, M.J.; Pradhan, D.; Bhargava, R.; Edwards, R.P.; Kelley, J.L.; Comerci, J.T.; Olawaiye, A.B.; Courtney-Brooks, M.B.; Bockmeier, M.M.; et al. Human papillomavirus infection mediates response and outcome of vulvar squamous cell carcinomas treated with radiation therapy. Gynecol. Oncol. 2018, 151, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Welzel, G.; Ottstadt, M.; Lohr, F.; Severa, S.; Prigge, E.S.; Wentzensen, N.; Trunk, M.J.; Wenz, F.; von Knebel-Doeberitz, M.; et al. Prognostic Relevance of HPV Infection and p16 Overexpression in Squamous Cell Anal Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 819–827. [Google Scholar] [CrossRef]
- Meulendijks, D.; Tomasoa, N.B.; Dewit, L.; Smits, P.H.; Bakker, R.; van Velthuysen, M.L.; Rosenberg, E.H.; Beijnen, J.H.; Schellens, J.H.; Cats, A. HPV-negative squamous cell carcinoma of the anal canal is unresponsive to standard treatment and frequently carries disruptive mutations in TP53. Br. J. Cancer 2015, 112, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Serup-Hansen, E.; Linnemann, D.; Skovrider-Ruminski, W.; Hogdall, E.; Geertsen, P.F.; Havsteen, H. Human papillomavirus genotyping and p16 expression as prognostic factors for patients with American Joint Committee on Cancer stages I to III carcinoma of the anal canal. J. Clin. Oncol. 2014, 32, 1812–1817. [Google Scholar] [CrossRef] [Green Version]
- Chera, B.S.; Amdur, R.J.; Green, R.; Shen, C.; Gupta, G.; Tan, X.; Knowles, M.; Fried, D.; Hayes, N.; Weiss, J.; et al. Phase II Trial of De-Intensified Chemoradiotherapy for Human Papillomavirus-Associated Oropharyngeal Squamous Cell Carcinoma. J. Clin. Oncol. 2019, 37, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Yom, S.S.; Torres-Saavedra, P.; Caudell, J.J.; Waldron, J.N.; Gillison, M.L.; Xia, P.; Truong, M.T.; Kong, C.; Jordan, R.; Subramaniam, R.M.; et al. Reduced-Dose Radiation Therapy for HPV-Associated Oropharyngeal Carcinoma (NRG Oncology HN002). J. Clin. Oncol. 2021, 39, 956–965. [Google Scholar] [CrossRef]
- Marur, S.; Li, S.; Cmelak, A.J.; Gillison, M.L.; Zhao, W.J.; Ferris, R.L.; Westra, W.H.; Gilbert, J.; Bauman, J.E.; Wagner, L.I.; et al. E1308: Phase II Trial of Induction Chemotherapy Followed by Reduced-Dose Radiation and Weekly Cetuximab in Patients With HPV-Associated Resectable Squamous Cell Carcinoma of the Oropharynx-ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2017, 35, 490–497. [Google Scholar] [CrossRef]
- Chen, A.M.; Felix, C.; Wang, P.C.; Hsu, S.; Basehart, V.; Garst, J.; Beron, P.; Wong, D.; Rosove, M.H.; Rao, S.; et al. Reduced-dose radiotherapy for human papillomavirus-associated squamous-cell carcinoma of the oropharynx: A single-arm, phase 2 study. Lancet Oncol. 2017, 18, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.J.; Price, K.A.; Moore, E.J.; Patel, S.H.; Hinni, M.L.; Garcia, J.J.; Graner, D.E.; Foster, N.R.; Ginos, B.; Neben-Wittich, M.; et al. Phase II Evaluation of Aggressive Dose De-Escalation for Adjuvant Chemoradiotherapy in Human Papillomavirus-Associated Oropharynx Squamous Cell Carcinoma. J. Clin. Oncol. 2019, 37, 1909–1918. [Google Scholar] [CrossRef]
- Nulton, T.J.; Olex, A.L.; Dozmorov, M.; Morgan, I.M.; Windle, B. Analysis of The Cancer Genome Atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 17684–17699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nulton, T.J.; Kim, N.K.; DiNardo, L.J.; Morgan, I.M.; Windle, B. Patients with integrated HPV16 in head and neck cancer show poor survival. Oral Oncol. 2018, 80, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [Green Version]
- Rieckmann, T.; Tribius, S.; Grob, T.J.; Meyer, F.; Busch, C.J.; Petersen, C.; Dikomey, E.; Kriegs, M. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother. Oncol. 2013, 107, 242–246. [Google Scholar] [CrossRef]
- Lilja-Fischer, J.K.; Ulhoi, B.P.; Alsner, J.; Stougaard, M.; Thomsen, M.S.; Busk, M.; Lassen, P.; Steiniche, T.; Nielsen, V.E.; Overgaard, J. Characterization and radiosensitivity of HPV-related oropharyngeal squamous cell carcinoma patient-derived xenografts. Acta Oncol. 2019, 58, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Nickel, K.P.; Torres, A.D.; Lee, D.; Lambert, P.F.; Kimple, R.J. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother. Oncol. 2014, 113, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Kranjc Brezar, S.; Prevc, A.; Niksic Zakelj, M.; Brozic, A.; Cemazar, M.; Strojan, P.; Sersa, G. Synergistic effect of cisplatin chemotherapy combined with fractionated radiotherapy regimen in HPV-positive and HPV-negative experimental pharyngeal squamous cell carcinoma. Sci. Rep. 2020, 10, 1563. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Taniguchi, C.M.; Klopp, A.H.; Colbert, L.E.; Lin, S.H.; Wang, L.; Frederick, M.J.; Osman, A.A.; Pickering, C.R.; Frank, S.J. Biology of the Radio- and Chemo-Responsiveness in HPV Malignancies. Semin. Radiat. Oncol. 2021, 31, 274–285. [Google Scholar] [CrossRef]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High-Risk Alphapapillomavirus Oncogenes Impair the Homologous Recombination Pathway. J. Virol. 2017, 91, e01084-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Ma, L.; Jones, T.; Palomero, L.; Pujana, M.A.; Martinez-Ruiz, H.; Ha, P.K.; Murnane, J.; Cuartas, I.; Seoane, J.; et al. Subjugation of TGFbeta Signaling by Human Papilloma Virus in Head and Neck Squamous Cell Carcinoma Shifts DNA Repair from Homologous Recombination to Alternative End Joining. Clin. Cancer Res. 2018, 24, 6001–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeman, J.E.; Li, Y.; Bell, A.; Hussain, S.S.; Majumdar, R.; Rong-Mullins, X.; Blecua, P.; Damerla, R.; Narang, H.; Ravindran, P.T.; et al. Human papillomavirus 16 promotes microhomology-mediated end-joining. Proc. Natl. Acad. Sci. USA 2019, 116, 21573–21579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitz, J.; Blanchet, S.A.; Gameiro, S.F.; Biquand, E.; Morgan, T.M.; Galloy, M.; Dessapt, J.; Lavoie, E.G.; Blondeau, A.; Smith, B.C.; et al. Human papillomavirus E7 oncoprotein targets RNF168 to hijack the host DNA damage response. Proc. Natl. Acad. Sci. USA 2019, 116, 19552–19562. [Google Scholar] [CrossRef] [Green Version]
- Prevc, A.; Kranjc, S.; Cemazar, M.; Todorovic, V.; Zegura, B.; Novak, M.; Filipic, M.; Flezar, M.S.; Kirbis, I.S.; Rotter, A.; et al. Dose-Modifying Factor of Radiation Therapy with Concurrent Cisplatin Treatment in HPV-Positive Squamous Cell Carcinoma: A Preclinical Study. Radiat. Res. 2018, 189, 644–651. [Google Scholar] [CrossRef]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 Regulates Transcription of the Topoisomerase IIbeta-Binding Protein 1 (TopBP1) Gene to Activate the ATR Pathway and Promote Human Papillomavirus Replication. mBio 2015, 6, e02006-15. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.L.; Macdonald, A. Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses 2020, 12, 977. [Google Scholar] [CrossRef]
- Dok, R.; Kalev, P.; Van Limbergen, E.J.; Asbagh, L.A.; Vazquez, I.; Hauben, E.; Sablina, A.; Nuyts, S. p16INK4a impairs homologous recombination-mediated DNA repair in human papillomavirus-positive head and neck tumors. Cancer Res. 2014, 74, 1739–1751. [Google Scholar] [CrossRef] [Green Version]
- Jirawatnotai, S.; Hu, Y.; Michowski, W.; Elias, J.E.; Becks, L.; Bienvenu, F.; Zagozdzon, A.; Goswami, T.; Wang, Y.E.; Clark, A.B.; et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 2011, 474, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, P.; Molkentine, D.P.; Chen, C.; Molkentine, J.M.; Piao, H.; Raju, U.; Zhang, J.; Valdecanas, D.R.; Tailor, R.C.; et al. TRIP12 as a mediator of human papillomavirus/p16-related radiation enhancement effects. Oncogene 2017, 36, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Busch, C.J.; Becker, B.; Kriegs, M.; Gatzemeier, F.; Kruger, K.; Mockelmann, N.; Fritz, G.; Petersen, C.; Knecht, R.; Rothkamm, K.; et al. Similar cisplatin sensitivity of HPV-positive and -negative HNSCC cell lines. Oncotarget 2016, 7, 35832–35842. [Google Scholar] [CrossRef]
- Hussain, S.S.; Majumdar, R.; Moore, G.M.; Narang, H.; Buechelmaier, E.S.; Bazil, M.J.; Ravindran, P.T.; Leeman, J.E.; Li, Y.; Jalan, M.; et al. Measuring nonhomologous end-joining, homologous recombination and alternative end-joining simultaneously at an endogenous locus in any transfectable human cell. Nucleic Acids Res. 2021, 49, e74. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [Green Version]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.; Laimins, L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio 2018, 9, e00064-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e923. [Google Scholar] [CrossRef] [Green Version]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Chappell, W.H.; Gautam, D.; Ok, S.T.; Johnson, B.A.; Anacker, D.C.; Moody, C.A. Homologous Recombination Repair Factors Rad51 and BRCA1 Are Necessary for Productive Replication of Human Papillomavirus 31. J. Virol. 2015, 90, 2639–2652. [Google Scholar] [CrossRef] [Green Version]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Kim, B.; Moody, C.A. HPV31 utilizes the ATR-Chk1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating Keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J. Virol. 2015, 89, 4980–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.; Bristol, M.L.; Smith, N.W.; James, C.D.; Wang, X.; Pichierri, P.; Morgan, I.M. Werner Helicase Control of Human Papillomavirus 16 E1-E2 DNA Replication Is Regulated by SIRT1 Deacetylation. mBio 2019, 10, e00263-19. [Google Scholar] [CrossRef] [Green Version]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [Green Version]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A Functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Gaykalova, D.A.; Guo, T.; Favorov, A.V.; Fertig, E.J.; Tamayo, P.; Callejas-Valera, J.L.; Allevato, M.; Gilardi, M.; Santos, J.; et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene 2020, 39, 6327–6339. [Google Scholar] [CrossRef] [PubMed]
- Pang, E.; Delic, N.C.; Hong, A.; Zhang, M.; Rose, B.R.; Lyons, J.G. Radiosensitization of oropharyngeal squamous cell carcinoma cells by human papillomavirus 16 oncoprotein E6 *I. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 860–865. [Google Scholar] [CrossRef]
- Shamseddine, A.A.; Burman, B.; Lee, N.Y.; Zamarin, D.; Riaz, N. Tumor Immunity and Immunotherapy for HPV-Related Cancers. Cancer Discov. 2021, 11, 1896–1912. [Google Scholar] [CrossRef]
- Cleary, C.; Leeman, J.E.; Higginson, D.S.; Katabi, N.; Sherman, E.; Morris, L.; McBride, S.; Lee, N.; Riaz, N. Biological Features of Human Papillomavirus-related Head and Neck Cancers Contributing to Improved Response. Clin. Oncol. R Coll. Radiol. 2016, 28, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar]
- White, A.E.; Livanos, E.M.; Tlsty, T.D. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev. 1994, 8, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Deriano, L.; Stracker, T.H.; Baker, A.; Petrini, J.H.; Roth, D.B. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol. Cell 2009, 34, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, W.; Weiler, I.J.; Schuler, A.; Phillips, R.A.; Rosenberg, N.; Mak, T.W.; Kearney, J.F.; Perry, R.P.; Bosma, M.J. Rearrangement of antigen receptor genes is defective in mice with severe combined immune deficiency. Cell 1986, 46, 963–972. [Google Scholar] [CrossRef]
- Riaz, N.; Sherman, E.; Pei, X.; Schoder, H.; Grkovski, M.; Paudyal, R.; Katabi, N.; Selenica, P.; Yamaguchi, T.N.; Ma, D.; et al. Precision Radiotherapy: Reduction in Radiation for Oropharyngeal Cancer in the 30 ROC Trial. J. Natl. Cancer Inst. 2021, 113, 742–751. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or Without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Spisak, S.; Jia, L.; Cronin, A.M.; Csabai, I.; Ledet, E.; Sartor, A.O.; Rainville, I.; O’Connor, E.P.; Herbert, Z.T.; et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 2017, 123, 3532–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.C.; Zhang, J.; Xu, B.H.; Cai, L.; Ragaz, J.; Wang, Z.H.; Wang, B.Y.; Teng, Y.E.; Tong, Z.S.; Pan, Y.Y.; et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–446. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Xie, X. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget 2017, 8, 97657–97670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermorken, J.B.; Psyrri, A.; Mesia, R.; Peyrade, F.; Beier, F.; de Blas, B.; Celik, I.; Licitra, L. Impact of tumor HPV status on outcome in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck receiving chemotherapy with or without cetuximab: Retrospective analysis of the phase III EXTREME trial. Ann. Oncol. 2014, 25, 801–807. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Double-strand break repair pathways and proposed mechanisms of HR-HPV mediated effects on pathway usage. Figure created with Biorender.com.
Figure 1.
Double-strand break repair pathways and proposed mechanisms of HR-HPV mediated effects on pathway usage. Figure created with Biorender.com.


{kind=link}
Table 1.
Comparison of genomic characteristics and therapy response in HPV positive and negative cancers.
Table 1.
Comparison of genomic characteristics and therapy response in HPV positive and negative cancers.
| Characteristic | HPV+ HNSCC | HPV− HNSCC |
|---|---|---|
| Dysregulated tumor suppressors | Rb (E7), p53 (E6) | p16(INK4A), p53 |
| Copy number alterations (median) | 113 | 136 |
| “M” class tumors | 58% | 27% |
| Somatic mutation frequency | Similar | Similar |
| Single base substitutions signature | APOBEC | Smoking |
| Alt-EJ genomic scars | Higher | Lower |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hussain, S.S.; Lundine, D.; Leeman, J.E.; Higginson, D.S. Genomic Signatures in HPV-Associated Tumors. Viruses 2021, 13, 1998. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101998
AMA Style
Hussain SS, Lundine D, Leeman JE, Higginson DS. Genomic Signatures in HPV-Associated Tumors. Viruses. 2021; 13(10):1998. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101998
Chicago/Turabian StyleHussain, Suleman S., Devon Lundine, Jonathan E. Leeman, and Daniel S. Higginson. 2021. "Genomic Signatures in HPV-Associated Tumors" Viruses 13, no. 10: 1998. https://0-doi-org.brum.beds.ac.uk/10.3390/v13101998
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.