
Transcriptomic Studies Suggest a Coincident Role for Apoptosis and Pyroptosis but Not for Autophagic Neuronal Death in TBEV-Infected Human Neuronal/Glial Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Culture of Human Neural Progenitor Cells
2.3. Neuronal and Glial Differentiation
2.4. Magnetic-Activated Cell Sorting
2.5. Virus and Infection
2.6. RNA Isolation and qPCR
2.7. RT2 Profiler PCR Array
2.8. Immunofluorescence Assays and Cell Enumeration
2.9. Statistical Analyses
3. Results
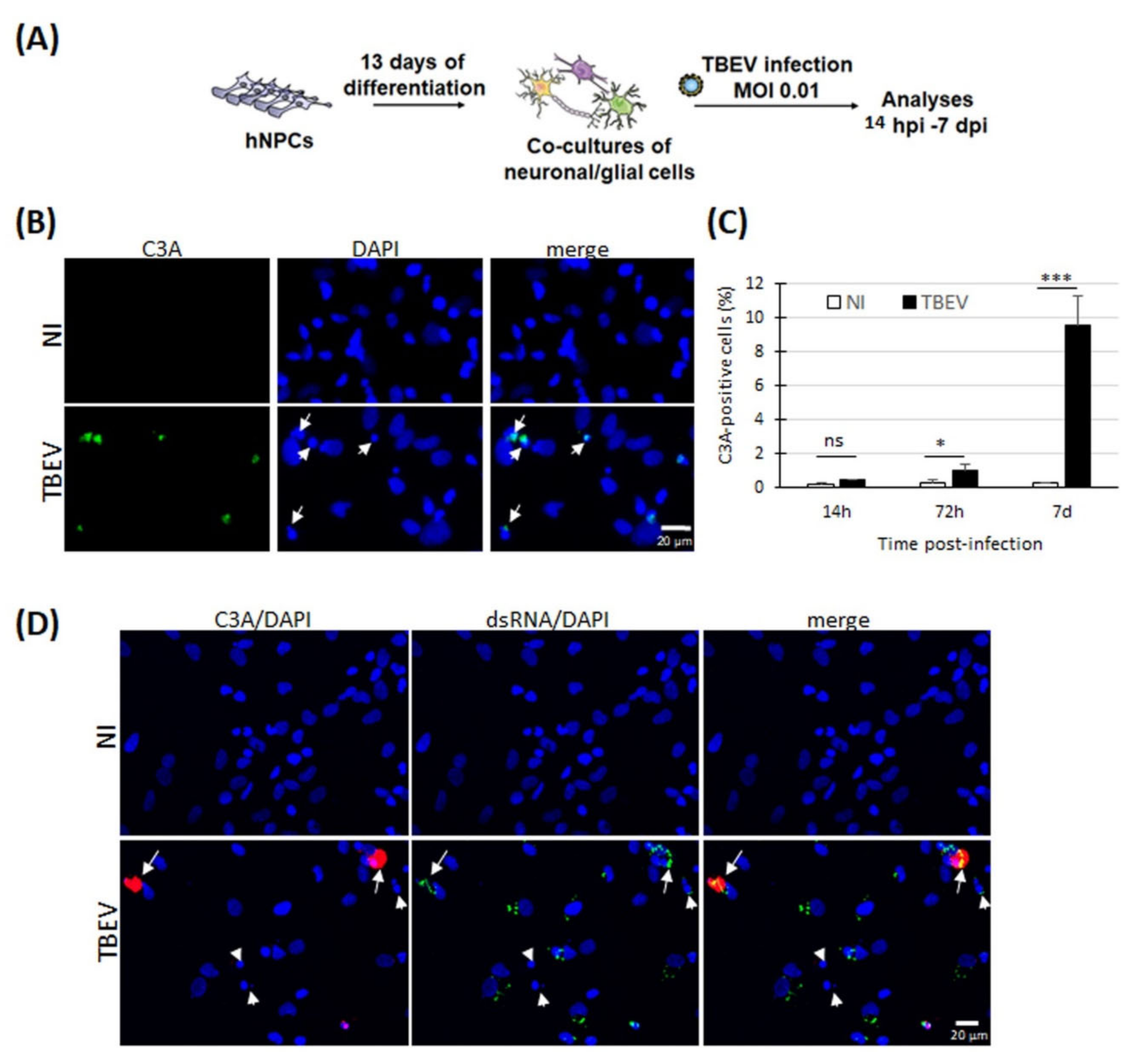
3.1. Evidence of Apoptotic Death in Human Neuronal/Glial Cells Infected with TBEV
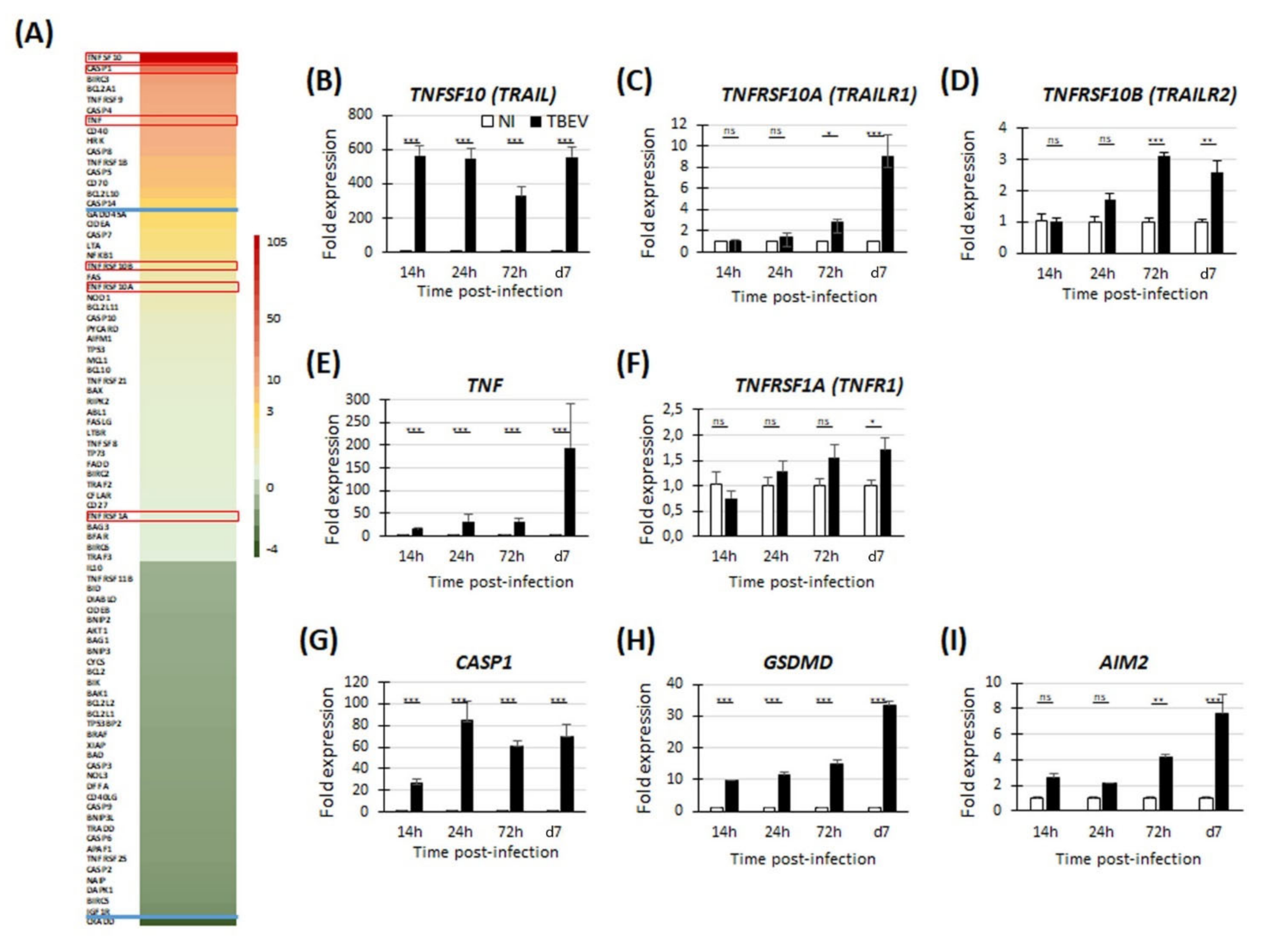
3.2. Transcriptomic Analyses Reveal Up-Regulation of Genes Involved in Apoptosis and Pyroptosis in TBEV-Infected Human Neuronal/Glial Cells
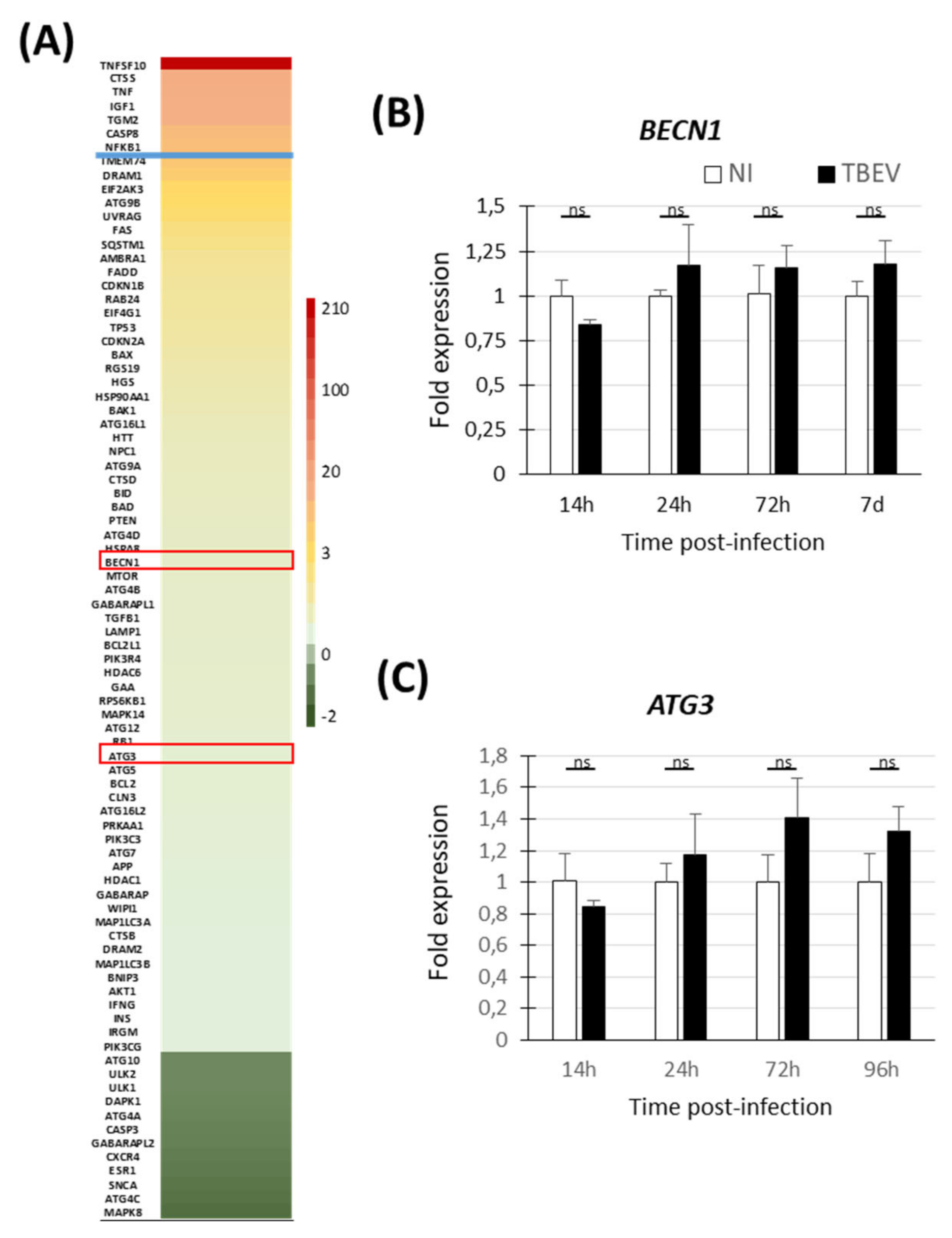
3.3. Transcriptomic Analyses Do Not Show Evidence of Activation of Autophagic Pathway in TBEV-Infected Human Neuronal/Glial Cells
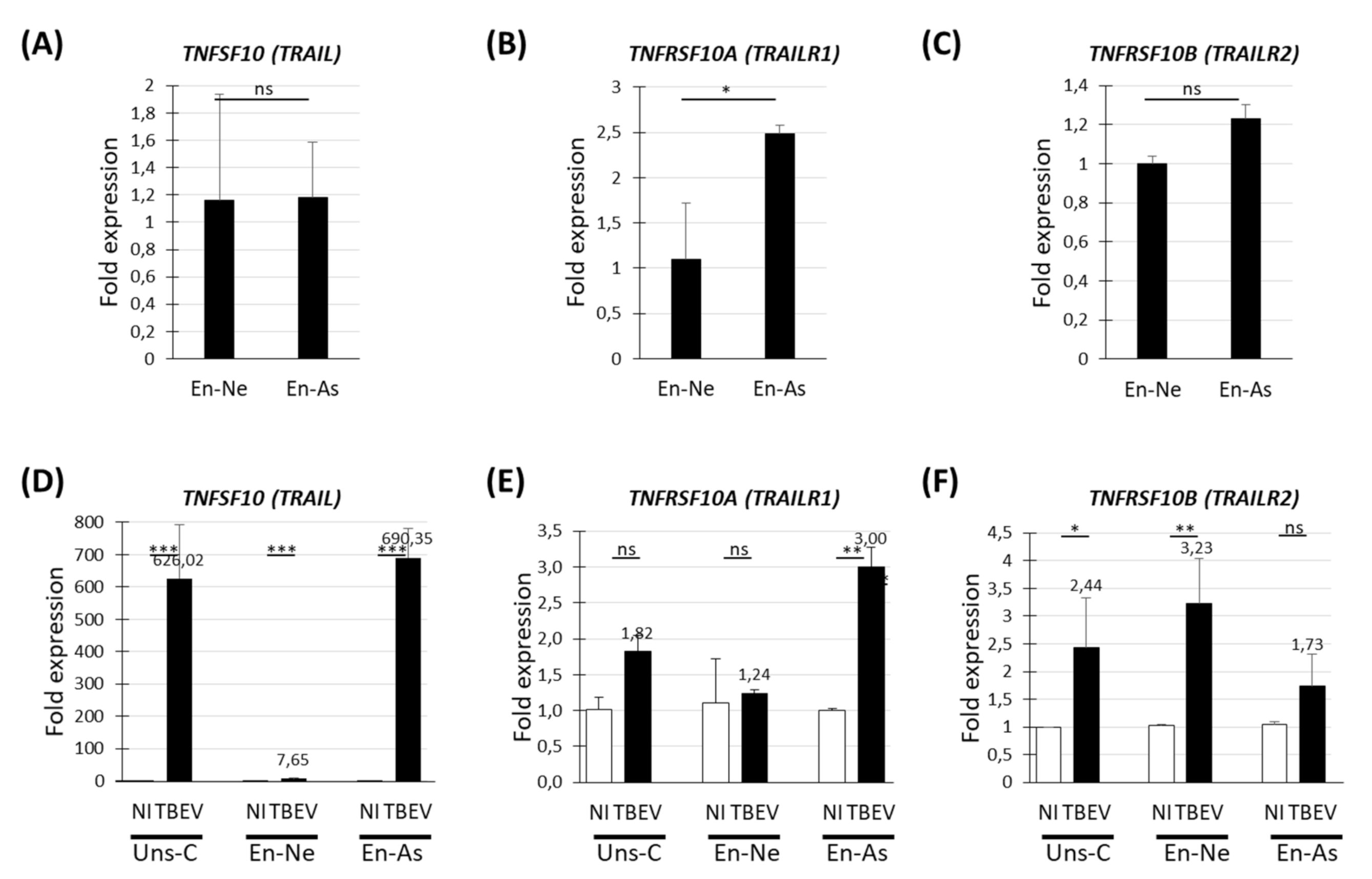
3.4. Human Neurons and Human Astrocytes Differentially Expressed the TNFSF10, TNFRSF10A and TNFRSF10B Genes in Basal and TBEV-Infected Conditions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beauté, J.; Spiteri, G.; Warns-Petit, E.; Zeller, H. Tick-borne encephalitis in Europe, 2012 to 2016. Eurosurveillance 2018, 23, 1800201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantke, O.D.; Escadafal, C.; Niedrig, M.; Pfeffer, M.; collective on behalf of the Working Group for Tick-Borne Encephalitis Virus. Tick-borne encephalitis in Europe, 2007 to 2009. Eurosurveillance 2011, 16, 19976. [Google Scholar] [CrossRef] [Green Version]
- Borde, J.P.; Kaier, K.; Hehn, P.; Matzarakis, A.; Frey, S.; Bestehorn, M.; Dobler, G.; Chitimia-Dobler, L. The complex interplay of climate, TBEV vector dynamics and TBEV infection rates in ticks—Monitoring a natural TBEV focus in Germany, 2009–2018. PLoS ONE 2021, 16, e0244668. [Google Scholar] [CrossRef]
- Ruzek, D.; Županc, T.A.; Borde, J.; Chrdle, A.; Eyer, L.; Karganova, G.; Kholodilov, I.; Knap, N.; Kozlovskaya, L.; Matveev, A.; et al. Tick-borne encephalitis in Europe and Russia: Review of pathogenesis, clinical features, therapy, and vaccines. Antivir. Res. 2019, 164, 23–51. [Google Scholar] [CrossRef]
- Balogh, Z.; Ferenczi, E.; Szeles, K.; Stefanoff, P.; Gut, W.; Szomor, K.N.; Takacs, M.; Berencsi, G. Tick-borne encephalitis outbreak in Hungary due to consumption of raw goat milk. J. Virol. Methods 2010, 163, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, S.; Oehme, R.; Buckenmaier, T.; Beer, M.; Jeffery-Smith, A.; Spannenkrebs, M.; Haag-Milz, S.; Wagner-Wiening, C.; Schlegel, C.; Fritz, J.; et al. A cluster of two human cases of tick-borne encephalitis (TBE) transmitted by unpasteurised goat milk and cheese in Germany, May 2016. Eurosurveillance 2018, 23, 17-00336. [Google Scholar] [CrossRef]
- Dorko, E.; Hockicko, J.; Rimárová, K.; Bušová, A.; Popaďák, P.; Popaďáková, J.; Schréter, I. Milk outbreaks of tick-borne encephalitis in Slovakia, 2012-2016. Cent. Eur. J. Public Health 2018, 26, S47–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formanova, P.P.; Palus, M.; Salat, J.; Hönig, V.; Stefanik, M.; Svoboda, P.; Ruzek, D. Changes in cytokine and chemokine profiles in mouse serum and brain, and in human neural cells, upon tick-borne encephalitis virus infection. J. Neuroinflamm. 2019, 16, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelpi, E.; Preusser, M.; Laggner, U.; Garzuly, F.; Holzmann, H.; Heinz, F.X.; Budka, H. Inflammatory response in human tick-borne encephalitis: Analysis of postmortem brain tissue. J. NeuroVirol. 2006, 12, 322–327. [Google Scholar] [CrossRef]
- Gelpi, E.; Preusser, M.; Garzuly, F.; Holzmann, H.; Heinz, F.X.; Budka, H. Visualization of Central European Tick-Borne Encephalitis Infection in Fatal Human Cases. J. Neuropathol. Exp. Neurol. 2005, 64, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Hayasaka, D.; Nagata, N.; Fujii, Y.; Hasegawa, H.; Sata, T.; Suzuki, R.; Gould, E.A.; Takashima, I.; Koike, S. Mortality following peripheral infection with Tick-borne encephalitis virus results from a combination of central nervous system pathology, systemic inflammatory and stress responses. Virology 2009, 390, 139–150. [Google Scholar] [CrossRef]
- Růžek, D.; Salát, J.; Palus, M.; Gritsun, T.S.; Gould, E.A.; Dyková, I.; Skallová, A.; Jelínek, J.; Kopecký, J.; Grubhoffer, L. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology 2009, 384, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Sims, J.; Botez, G.; Winckler, J. Spatial resolution of phospholipid scramblase 1 (PLSCR1), caspase-3 activation and DNA-fragmentation in the human hippocampus after cerebral ischemia. Neurochem. Int. 2003, 43, 79–87. [Google Scholar] [CrossRef]
- Jellinger, K.A. Cell death mechanisms in neurodegeneration. J. Cell. Mol. Med. 2001, 5, 1–17. [Google Scholar] [CrossRef]
- Stadelmann, C.; Deckwerth, T.L.; Srinivasan, A.; Bancher, C.; Brück, W.; Jellinger, K.; Lassmann, H. Activation of Caspase-3 in Single Neurons and Autophagic Granules of Granulovacuolar Degeneration in Alzheimer’s Disease: Evidence for Apoptotic Cell Death. Am. J. Pathol. 1999, 155, 1459–1466. [Google Scholar] [CrossRef]
- Henshall, D.C.; Bonislawski, D.P.; Skradski, S.L.; Araki, T.; Lan, J.-Q.; Schindler, C.K.; Meller, R.; Simon, R.P. Formation of the Apaf-1/cytochrome c complex precedes activation of caspase-9 during seizure-induced neuronal death. Cell Death Differ. 2001, 8, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.C.; Davis, R.P.; Bond, B.C.; Campbell, C.A.; James, M.F.; Parsons, A.A.; Philpott, K.L. Caspase mRNA expression in a rat model of focal cerebral ischemia. Mol. Brain Res. 2001, 89, 133–146. [Google Scholar] [CrossRef]
- Chu, J.J.H.; Ng, M.L. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J. Gen. Virol. 2003, 84, 3305–3314. [Google Scholar] [CrossRef]
- Ho, C.-Y.; Ames, H.M.; Tipton, A.; Vezina, G.; Liu, J.S.; Scafidi, J.; Torii, M.; Rodriguez, F.J.; Du Plessis, A.; DeBiasi, R.L. Differential neuronal susceptibility and apoptosis in congenital Zika virus infection. Ann. Neurol. 2017, 82, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Isaeva, M.P.; Leonova, G.N.; Kozhemiako, V.B.; Borisevich, V.G.; Maĭstrovskaia, O.S.; A Rasskazov, V. Apoptosis as a mechanism for the cytopathic action of tick-borne encephalitis virus. Vopr. Virusol. 1998, 43, 182–186. [Google Scholar]
- Kamalov, N.I.; Novozhilova, A.P.; Kreĭchman, G.S.; Sokolova, E.D. The morphological characteristics of cell death in different forms of acute tick-borne encephalitis. Morfology 1998, 114, 54–58. [Google Scholar]
- Ruzek, D.; Vancová, M.; Tesařová, M.; Ahantarig, A.; Kopecký, J.; Grubhoffer, L. Morphological changes in human neural cells following tick-borne encephalitis virus infection. J. Gen. Virol. 2009, 90, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.; Cochet-Bernoin, M.; Gonzalez, G.; Montero-Menei, C.N.; Blanchet, O.; Benchoua, A.; Boissart, C.; Lecollinet, S.; Richardson, J.; Haddad, N.; et al. Pathological modeling of TBEV infection reveals differential innate immune responses in human neurons and astrocytes that correlate with their susceptibility to infection. J. Neuroinflamm. 2020, 17, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, B.; Dixit, V.M.; Power, C. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci. 2020, 43, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-C.; Zhang, J.-G.; Tan, M.-S.; Chen-Chen, T.; Meng, D.; Jiang, T.; Meng, X.-F.; Li, Y.; Sun, Z.; Li, M.-M.; et al. NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model. J. Neuroinflamm. 2015, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.-S.; Tan, L.; Jiang, T.; Zhu, X.-C.; Wang, H.-F.; Jia, C.-D.; Yu, J.-T. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef]
- Lim, S.M.; Ham, H.-J.V.D.; Oduber, M.; Martina, E.; Zaaraoui-Boutahar, F.; Roose, J.M.; van Ijcken, W.; Osterhaus, A.D.M.E.; Andeweg, A.C.; Koraka, P.; et al. Transcriptomic Analyses Reveal Differential Gene Expression of Immune and Cell Death Pathways in the Brains of Mice Infected with West Nile Virus and Chikungunya Virus. Front. Microbiol. 2017, 8, 1556. [Google Scholar] [CrossRef]
- Koraka, P.; Martina, B.E.; Smreczak, M.; Orlowska, A.; Marzec-Grządziel, A.; Trebas, P.; Roose, J.M.; Begeman, L.; Gerhauser, I.; Wohlsein, P.; et al. Inhibition of caspase-1 prolongs survival of mice infected with rabies virus. Vaccine 2019, 37, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Matsunaga, S.; Jeremiah, S.S.; Nishi, M.; Miyakawa, K.; Morita, T.; Khatun, H.; Shimizu, H.; Okabe, N.; Kimura, H.; et al. Zika virus protease induces caspase-independent pyroptotic cell death by directly cleaving gasdermin D. Biochem. Biophys. Res. Commun. 2021, 534, 666–671. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Vicencio, J.M.; Kepp, O.; Joza, N.; Tajeddine, N.; Kroemer, G. Life, death and burial: Multifaceted impact of autophagy. Biochem. Soc. Trans. 2008, 36, 786–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakpoor, A.; Panyasrivanit, M.; Wikan, N.; Smith, D.R. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 2009, 90, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Linking dengue virus entry and translation/replication through amphisomes. Autophagy 2009, 5, 434–435. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Orba, Y.; Yamaguchi, H.; Takahashi, K.; Sasaki, M.; Hasebe, R.; Kimura, T.; Sawa, H. Autophagy inhibits viral genome replication and gene expression stages in West Nile virus infection. Virus Res. 2014, 191, 83–91. [Google Scholar] [CrossRef]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bily, T.; Palus, M.; Eyer, L.; Elsterová, J.; Vancová, M.; Růžek, D. Electron Tomography Analysis of Tick-Borne Encephalitis Virus Infection in Human Neurons. Sci. Rep. 2015, 5, 10745. [Google Scholar] [CrossRef] [Green Version]
- Brnic, D.; Stevanovic, V.; Cochet, M.; Agier, C.; Richardson, J.; Montero-Menei, C.N.; Milhavet, O.; Eloit, M.; Coulpier, M. Borna Disease Virus Infects Human Neural Progenitor Cells and Impairs Neurogenesis. J. Virol. 2011, 86, 2512–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scordel, C.; Huttin, A.; Cochet-Bernoin, M.; Szelechowski, M.; Poulet, A.; Richardson, J.; Benchoua, A.; Gonzalez-Dunia, D.; Eloit, M.; Coulpier, M. Borna Disease Virus Phosphoprotein Impairs the Developmental Program Controlling Neurogenesis and Reduces Human GABAergic Neurogenesis. PLoS Pathog. 2015, 11, e1004859. [Google Scholar] [CrossRef]
- Wallner, G.; Mandl, C.W.; Ecker, M.; Holzmann, H.; Stiasny, K.; Kunz, C.; Heinz, F.X. Characterization and complete genome sequences of high- and low-virulence variants of tick-borne encephalitis virus. J. Gen. Virol. 1996, 77, 1035–1042. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Charriaut-Marlangue, C.; Ben-Ari, Y. A Cautionary Note on the Use of the TUNEL Stain to Determine Apoptosis. Neuroreport 1995, 7, 61–64. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, M.P.; Chambers, J.A.; Pankhong, P.; Chattergoon, M.; Attatippaholkun, W.; Dang, K.; Shah, N.; Weiner, D.B. Host cell killing by the West Nile Virus NS2B–NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology 2006, 345, 56–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parquet, M.D.C.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, F.C.; Arvin, B.; White, R.F.; Miller, A.; Webb, C.L.; Willette, R.N.; Lysko, P.G.; Feuerstein, G.Z. Tumor Necrosis Factor-α. Stroke 1997, 28, 1233–1244. [Google Scholar] [CrossRef]
- Zhao, X.; Bausano, B.; Pike, B.R.; Newcomb-Fernandez, J.K.; Wang, K.; Shohami, E.; Ringger, N.; DeFord, S.; Anderson, D.K.; Hayes, R.L. TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J. Neurosci. Res. 2001, 64, 121–131. [Google Scholar] [CrossRef]
- Zhang, B.; Patel, J.; Croyle, M.; Diamond, M.S.; Klein, R.S. TNF-α-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J. Neuroimmunol. 2010, 224, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Verma, S.; Nerurkar, V.R. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflamm. 2010, 7, 73. [Google Scholar] [CrossRef] [Green Version]
- Dörr, J.; Roth, K.; Zurbuchen, U.; Deisz, R.; Bechmann, I.; Lehmann, T.-N.; Meier, S.; Nitsch, R.; Zipp, F. Tumor-necrosis-factor-related apoptosis-inducing-ligand (TRAIL)-mediated death of neurons in living human brain tissue is inhibited by flupirtine-maleate. J. Neuroimmunol. 2005, 167, 204–209. [Google Scholar] [CrossRef]
- Nitsch, R.; Bechmann, I.; A Deisz, R.; Haas, D.; Lehmann, T.N.; Wendling, U.; Zipp, F. Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 2000, 356, 827–828. [Google Scholar] [CrossRef]
- Aktas, O.; Smorodchenko, A.; Brocke, S.; Infante-Duarte, C.; Topphoff, U.S.; Vogt, J.; Prozorovski, T.; Meier, S.; Osmanova, V.; Pohl, E.; et al. Neuronal Damage in Autoimmune Neuroinflammation Mediated by the Death Ligand TRAIL. Neuron 2005, 46, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kichev, A.; Rousset, C.I.; Baburamani, A.; Levison, S.; Wood, T.; Gressens, P.; Thornton, C.; Hagberg, H. Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL) Signaling and Cell Death in the Immature Central Nervous System after Hypoxia-Ischemia and Inflammation. J. Biol. Chem. 2014, 289, 9430–9439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Erdmann, N.; Zhao, J.; Zheng, J. The signaling and apoptotic effects of TNF-related apoptosis-inducing ligand in HIV-1 associated dementia. Neurotox. Res. 2005, 8, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Trapp, B.D. Taking Two TRAILS. Neuron 2005, 46, 355–356. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhao, R.; Gao, K.; Wei, Z.; Yin, M.Y.; Lau, L.T.; Chui, D.; Yu, A.C.H. Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 67–80. [Google Scholar] [CrossRef]
- Bian, P.; Zheng, X.; Wei, L.; Ye, C.; Fan, H.; Cai, Y.; Zhang, Y.; Zhang, F.; Jia, Z.; Lei, Y. MLKL Mediated Necroptosis Accelerates JEV-Induced Neuroinflammation in Mice. Front. Microbiol. 2017, 8, 303. [Google Scholar] [CrossRef] [Green Version]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- He, Z.; An, S.; Chen, J.; Zhang, S.; Tan, C.; Yu, J.; Ye, H.; Wu, Y.; Yuan, J.; Wu, J.; et al. Neural progenitor cell pyroptosis contributes to Zika virus-induced brain atrophy and represents a therapeutic target. Proc. Natl. Acad. Sci. USA 2020, 117, 23869–23878. [Google Scholar] [CrossRef]
- de Sousa, J.R.; Azevedo, R.D.S.D.S.; Filho, A.J.M.; de Araujo, M.T.F.; Cruz, E.D.R.M.; Vasconcelos, B.C.B.; Cruz, A.C.R.; de Oliveira, C.S.; Martins, L.C.; Vasconcelos, B.H.B.; et al. In situ inflammasome activation results in severe damage to the central nervous system in fatal Zika virus microcephaly cases. Cytokine 2018, 111, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.; Gyeltshen, S.; Chaijaroenkul, W.; Na-Bangchang, K. Significance of Autophagy in Dengue Virus Infection: A Brief Review. Am. J. Trop. Med. Hyg. 2019, 100, 783–790. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fares, M.; Gorna, K.; Berry, N.; Cochet-Bernoin, M.; Piumi, F.; Blanchet, O.; Haddad, N.; Richardson, J.; Coulpier, M. Transcriptomic Studies Suggest a Coincident Role for Apoptosis and Pyroptosis but Not for Autophagic Neuronal Death in TBEV-Infected Human Neuronal/Glial Cells. Viruses 2021, 13, 2255. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112255
Fares M, Gorna K, Berry N, Cochet-Bernoin M, Piumi F, Blanchet O, Haddad N, Richardson J, Coulpier M. Transcriptomic Studies Suggest a Coincident Role for Apoptosis and Pyroptosis but Not for Autophagic Neuronal Death in TBEV-Infected Human Neuronal/Glial Cells. Viruses. 2021; 13(11):2255. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112255
Chicago/Turabian StyleFares, Mazigh, Kamila Gorna, Noémie Berry, Marielle Cochet-Bernoin, François Piumi, Odile Blanchet, Nadia Haddad, Jennifer Richardson, and Muriel Coulpier. 2021. "Transcriptomic Studies Suggest a Coincident Role for Apoptosis and Pyroptosis but Not for Autophagic Neuronal Death in TBEV-Infected Human Neuronal/Glial Cells" Viruses 13, no. 11: 2255. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112255