
A Combination of M50I and V151I Polymorphic Mutations in HIV-1 Subtype B Integrase Results in Defects in Autoprocessing

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. HIV RNA Sequencing
2.3. Sequence Assembly
2.4. Cells
2.5. Construction of Plasmids Encoding HIV Variants
2.6. Recombinant HIV-1 Viruses
2.7. HIV Replication Assay
2.8. Western Blotting
2.9. Homology Modeling of HIV-1 Integrase
2.10. Statistical Analysis
3. Results
3.1. Population Analysis of M50I in NIAID Clinical Samples
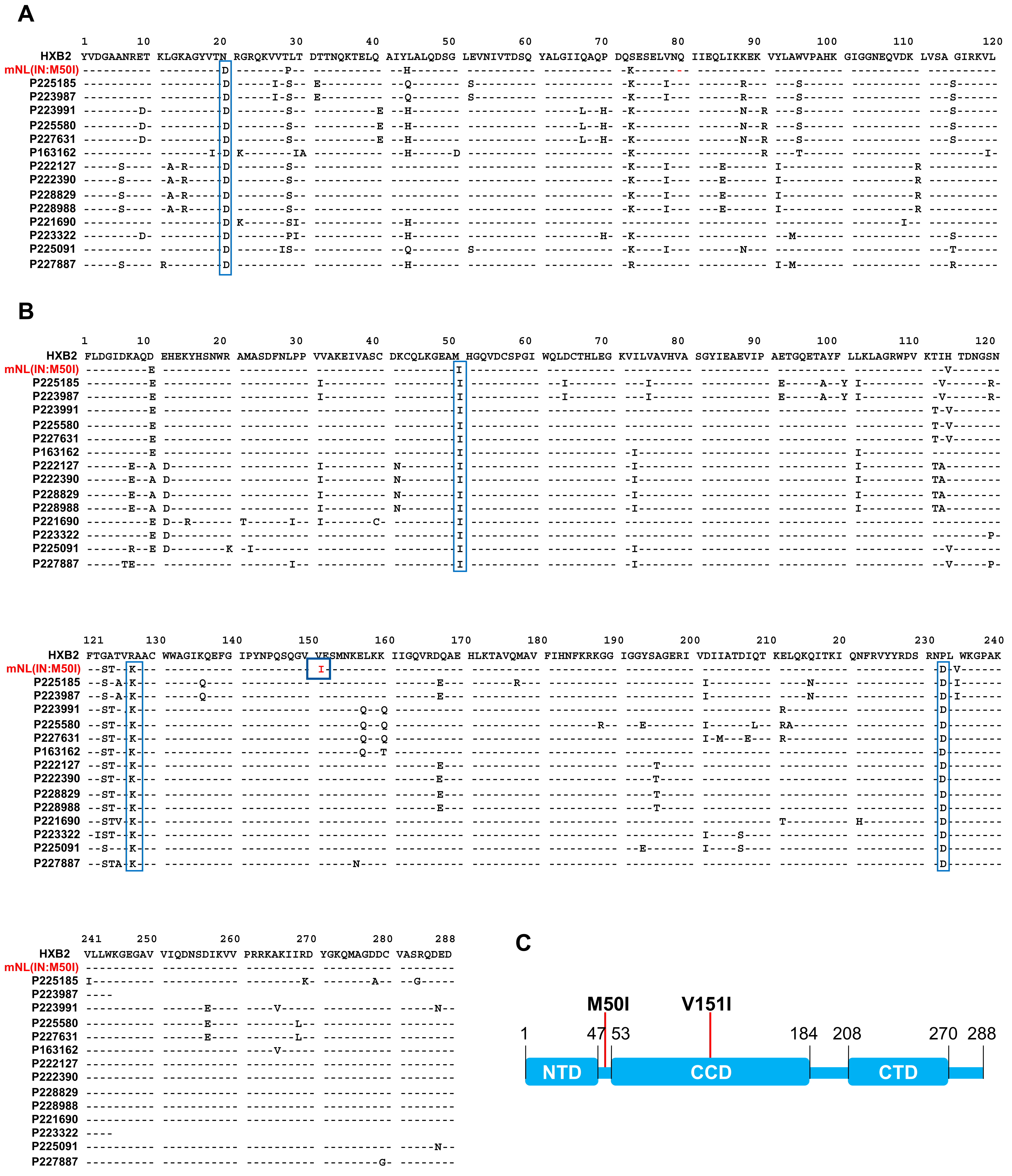
3.2. Analysis of Sequence Alignments
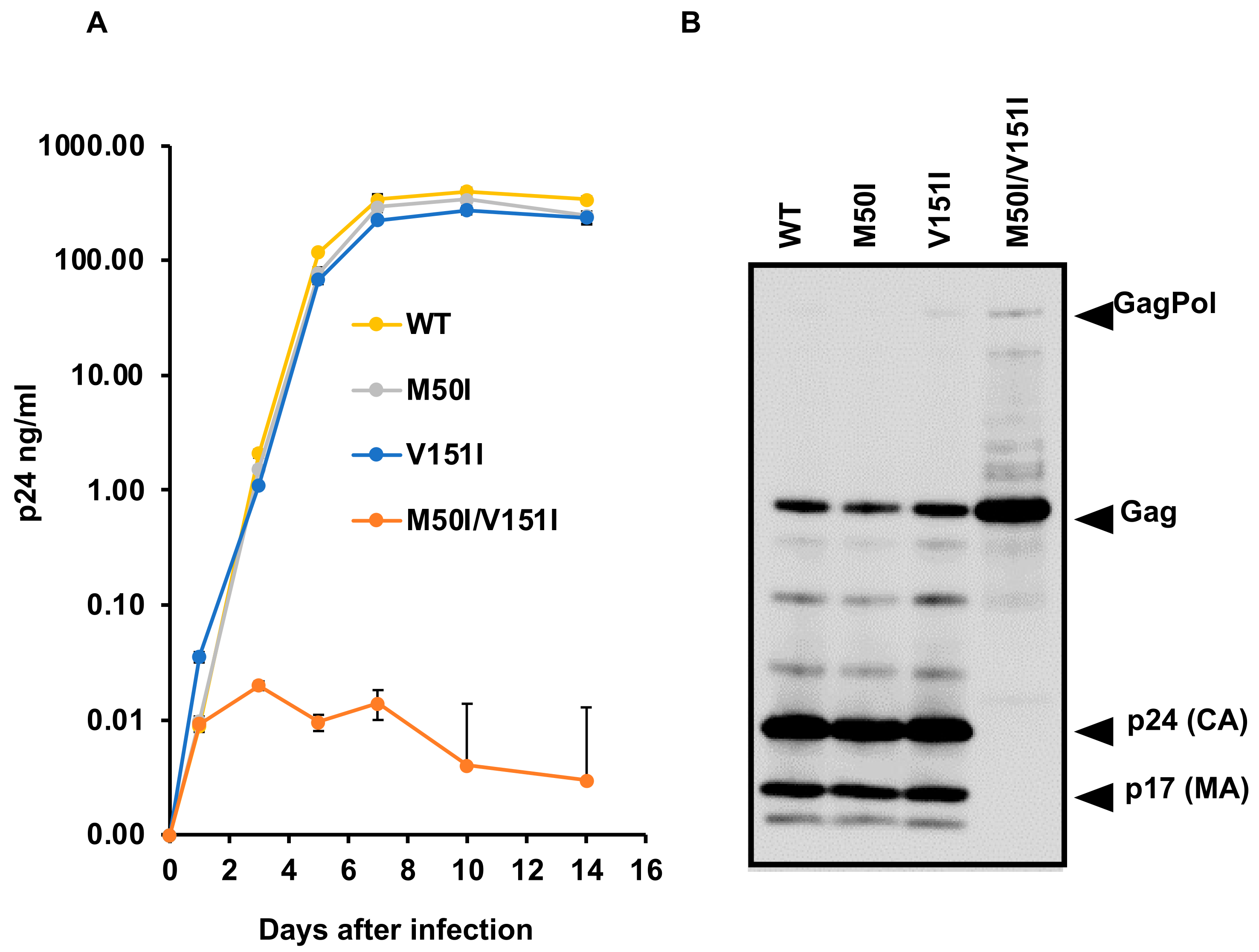
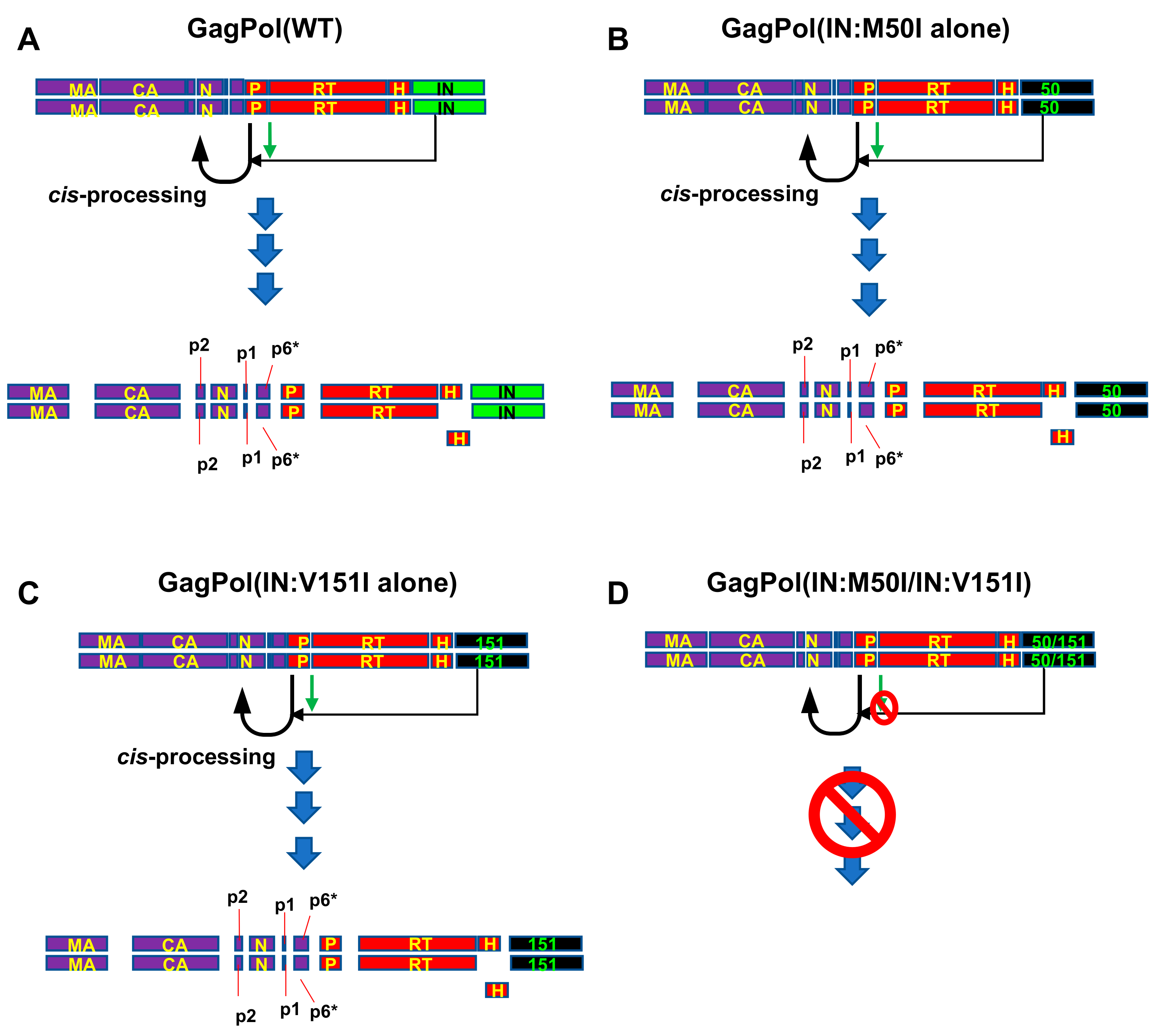
3.3. Evaluation of the Impact of IN:V151I Mutation on HIV Replication and Autoprocessing
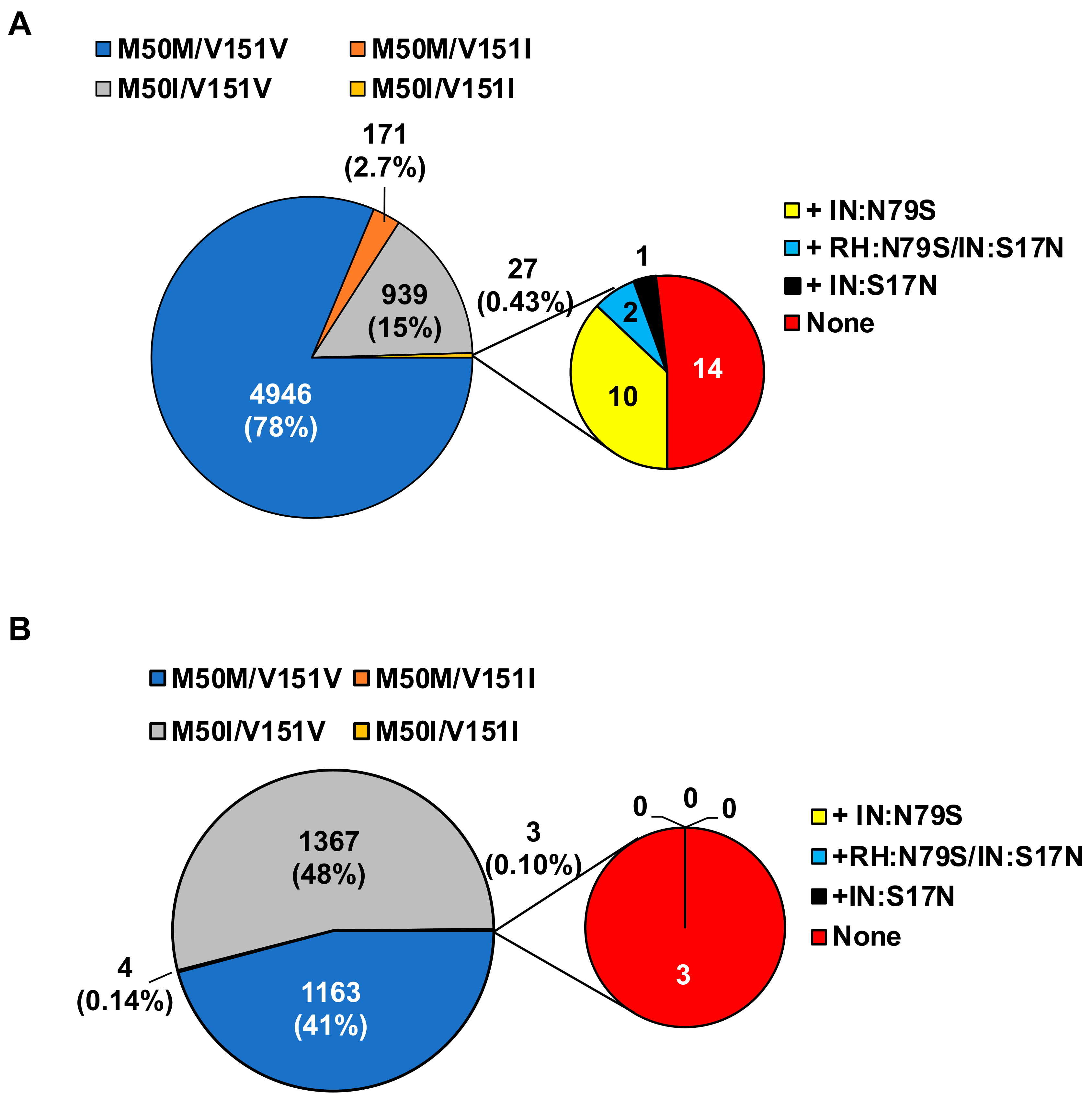
3.4. Population Analysis of the Combination of IN:M50I/IN:V151I in HIV Sequence Database
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALLINIs | allosteric IN inhibitors |
| ART | anti-retroviral therapy |
| CA | capsid protein |
| DTG | dolutegravir |
| FBS | fetal bovine serum |
| GWAS | Genome-wide association study |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HIV-1 | human immunodeficiency virus type 1 |
| IN | integrase |
| IN:M50I | methionine-to-isoleucine change at codon 50 of integrase |
| IN:S17N | serin-to-asparagine change at codon 17 of integrase |
| IN:V151 | valine-to-isoleucine change at codon 151of integrase |
| LANL | Los Alamos National Laboratory |
| MA | matrix protein |
| mNL | modified cloned HIVNL4.3 |
| NC | nucleocapsid protein |
| NGS | next generation sequence |
| NIAID | National Institute of Allergy and Infectious Diseases |
| NOPs | naturally occurring polymorphisms |
| PBMC | peripheral blood mononuclear cells |
| PHA | phytohemagglutinin |
| PR | protease |
| RH | RNaseH |
| RH:N79S | asparagine-to-serin change at codon 79 of RNaseH |
| RP10 | RPMI-1640 supplemented with 10% FBS |
| RT | reverse transcriptase |
| VL | virus load |
| WB | Western Blotting |
| WT | wild-type |
References
- Swanstrom, R.; Wills, J.W. Synthesis, Assembly, and Processing of Viral Proteins. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Freund, J.; Kellner, R.; Konvalinka, J.; Wolber, V.; Kräusslich, H.G.; Kalbitzer, H.R. A possible regulation of negative factor (Nef) activity of human immunodeficiency virus type 1 by the viral protease. Eur. J. Biochem. 1994, 223, 589–593. [Google Scholar] [CrossRef]
- Gaedigk-Nitschko, K.; Schön, A.; Wachinger, G.; Erfle, V.; Kohleisen, B. Cleavage of recombinant and cell derived human immunodeficiency virus 1 (HIV-1) Nef protein by HIV-1 protease. FEBS Lett. 1995, 357, 275–278. [Google Scholar] [CrossRef] [Green Version]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Everitt, L.E.; Choudhury, S.; Dunn, B.M.; Kaplan, A.H. Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism. J. Virol. 2004, 78, 8477–8485. [Google Scholar] [CrossRef] [Green Version]
- Louis, J.M.; Clore, G.M.; Gronenborn, A.M. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 1999, 6, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, Y.; Chen, C. Flexible catalytic site conformations implicated in modulation of HIV-1 protease autoprocessing reactions. Retrovirology 2011, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.A.; Soule, E.E.; Davidoff, K.S.; Daniels, S.I.; Naiman, N.E.; Yarchoan, R. Activity of human immunodeficiency virus type 1 protease inhibitors against the initial autocleavage in Gag-Pol polyprotein processing. Antimicrob Agents Chemother 2012, 56, 3620–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, A.; Moore, K.L.; Mak, J.; Sluis-Cremer, N.; de Bethune, M.P.; Tachedjian, G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006, 2, e119. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Cajas, J.L.; Pant-Pai, N.; Klein, M.B.; Wainberg, M.A. Role of genetic diversity amongst HIV-1 non-B subtypes in drug resistance: A systematic review of virologic and biochemical evidence. Aids Rev. 2008, 10, 212–223. [Google Scholar]
- Low, A.; Prada, N.; Topper, M.; Vaida, F.; Castor, D.; Mohri, H.; Hazuda, D.; Muesing, M.; Markowitz, M. Natural polymorphisms of human immunodeficiency virus type 1 integrase and inherent susceptibilities to a panel of integrase inhibitors. Antimicrob. Agents Chemother. 2009, 53, 4275–4282. [Google Scholar] [CrossRef] [Green Version]
- Ceccherini-Silberstein, F.; Malet, I.; Fabeni, L.; Dimonte, S.; Svicher, V.; D’Arrigo, R.; Artese, A.; Costa, G.; Bono, S.; Alcaro, S.; et al. Specific HIV-1 integrase polymorphisms change their prevalence in untreated versus antiretroviral-treated HIV-1-infected patients, all naive to integrase inhibitors. J. Antimicrob. Chemother. 2010, 65, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, M.; Chiarella, J.; Kozal, M.J. Natural polymorphism of the HIV-1 integrase gene and mutations associated with integrase inhibitor resistance. Antivir. Ther. 2007, 12, 563–570. [Google Scholar]
- Lundgren, J.D.; Babiker, A.G.; Gordin, F.; Emery, S.; Grund, B.; Sharma, S.; Avihingsanon, A.; Cooper, D.A.; Fätkenheuer, G.; Llibre, J.M.; et al. Initiation of Antiretroviral Therapy in Early Asymptomatic HIV Infection. N. Engl. J. Med. 2015, 373, 795–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielaite, M.; Zucco, A.G.; Bennedbaek, M.; Ekenberg, C.; Kan, V.L.; Touloumi, G.; Vandekerckhove, L.; Turner, D.; Neaton, J.; Lane, H.C.; et al. Association of HIV and host genetic variants in antiretroviral therapy-naive persons. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 8–11 March 2020. [Google Scholar]
- Imamichi, T.; Bernbaum, J.G.; Laverdure, S.; Yang, J.; Chen, Q.; Highbarger, H.; Hao, M.; Sui, H.; Dewar, R.; Chang, W.; et al. Natural Occurring Polymorphisms in HIV-1 Integrase and RNase H Regulate Viral Release and Autoprocessing. J. Virol. 2021, 95, e0132321. [Google Scholar] [CrossRef] [PubMed]
- Wares, M.; Mesplède, T.; Quashie, P.K.; Osman, N.; Han, Y.; Wainberg, M.A. The M50I polymorphic substitution in association with the R263K mutation in HIV-1 subtype B integrase increases drug resistance but does not restore viral replicative fitness. Retrovirology 2014, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migueles, S.A.; Chairez, C.; Lin, S.; Gavil, N.V.; Rosenthal, D.M.; Pooran, M.; Natarajan, V.; Rupert, A.; Dewar, R.; Rehman, T.; et al. Adoptive lymphocyte transfer to an HIV-infected progressor from an elite controller. JCI Insight 2019, 4, e130664. [Google Scholar] [CrossRef] [Green Version]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakruddin, J.M.; Lempicki, R.A.; Gorelick, R.J.; Yang, J.; Adelsberger, J.W.; Garcia-Pineres, A.J.; Pinto, L.A.; Lane, H.C.; Imamichi, T. Noninfectious papilloma virus-like particles inhibit HIV-1 replication: Implications for immune control of HIV-1 infection by IL-27. Blood 2007, 109, 1841–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [Green Version]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Brann, T.W.; Dewar, R.L.; Jiang, M.K.; Shah, A.; Nagashima, K.; Metcalf, J.A.; Falloon, J.; Lane, H.C.; Imamichi, T. Functional correlation between a novel amino acid insertion at codon 19 in the protease of human immunodeficiency virus type 1 and polymorphism in the p1/p6 Gag cleavage site in drug resistance and replication fitness. J. Virol. 2006, 80, 6136–6145. [Google Scholar] [CrossRef] [Green Version]
- Imamichi, T.; Berg, S.C.; Imamichi, H.; Lopez, J.C.; Metcalf, J.A.; Falloon, J.; Lane, H.C. Relative replication fitness of a high-level 3’-azido-3’-deoxythymidine-resistant variant of human immunodeficiency virus type 1 possessing an amino acid deletion at codon 67 and a novel substitution (Thr-->Gly) at codon 69. J. Virol. 2000, 74, 10958–10964. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Jaroszewski, L.; Li, Z.; Godzik, A. AIDA: Ab initio domain assembly server. Nucleic Acids Res. 2014, 42, W308–W313. [Google Scholar] [CrossRef] [Green Version]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal Structure of the Catalytic Domain of HIV-1 Integrase: Similarity to Other Polynucleotidyl Transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Craigie, R. HIV Integrase Structure and Function. In Advances in Virus Research; Rlaramorosch, K., Murphy, F.A., Shawn, A.J., Eds.; Academic Press: Cambridge, MA, USA, 1999; Volume 52, pp. 319–333. [Google Scholar]
- Wang, J.Y.; Ling, H.; Yang, W.; Craigie, R. Structure of a two-domain fragment of HIV-1 integrase: Implications for domain organization in the intact protein. Embo J. 2001, 20, 7333–7343. [Google Scholar] [CrossRef] [Green Version]
- Marchand, C.; Johnson, A.A.; Semenova, E.; Pommier, Y. Mechanisms and inhibition of HIV integration. Drug Discov. Today Dis. Mech. 2006, 3, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefic, K.; Salmona, M.; Capitao, M.; Splittgerber, M.; Maakaroun-Vermesse, Z.; Néré, M.L.; Bernard, L.; Chaix, M.L.; Barin, F.; Delaugerre, C. Unravelling the dynamics of selection of multiresistant variants to integrase inhibitors in an HIV-1-infected child using ultra-deep sequencing. J. Antimicrob. Chemother. 2017, 72, 850–854. [Google Scholar] [CrossRef]
- Ndashimye, E.; Li, Y.; Reyes, P.S.; Avino, M.; Olabode, A.S.; Kityo, C.M.; Kyeyune, F.; Nankya, I.; Quiñones-Mateu, M.E.; Barr, S.D.; et al. High-level resistance to bictegravir and cabotegravir in subtype A- and D-infected HIV-1 patients failing raltegravir with multiple resistance mutations. J. Antimicrob. Chemother. 2021, 76, 2965–2974. [Google Scholar] [CrossRef] [PubMed]
- Quashie, P.K.; Mesplède, T.; Han, Y.S.; Oliveira, M.; Singhroy, D.N.; Fujiwara, T.; Underwood, M.R.; Wainberg, M.A. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J. Virol. 2012, 86, 2696–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Nakahara, K.; Seki, T.; Miki, S.; Kawauchi, S.; Suyama, A.; Wakasa-Morimoto, C.; Kodama, M.; Endoh, T.; Oosugi, E.; et al. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antiviral. Res. 2008, 80, 213–222. [Google Scholar] [CrossRef]
- Blanco, J.L.; Varghese, V.; Rhee, S.Y.; Gatell, J.M.; Shafer, R.W. HIV-1 integrase inhibitor resistance and its clinical implications. J. Infect. Dis. 2011, 203, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Theys, K.; Libin, P.J.K.; Van Laethem, K.; Abecasis, A.B. An Evolutionary Model-Based Approach To Quantify the Genetic Barrier to Drug Resistance in Fast-Evolving Viruses and Its Application to HIV-1 Subtypes and Integrase Inhibitors. Antimicrob. Agents Chemother. 2019, 63, e00539-19. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.Y.; Liu, T.F.; Kiuchi, M.; Zioni, R.; Gifford, R.J.; Holmes, S.P.; Shafer, R.W. Natural variation of HIV-1 group M integrase: Implications for a new class of antiretroviral inhibitors. Retrovirology 2008, 5, 74. [Google Scholar] [CrossRef] [Green Version]
- Varghese, V.; Liu, T.F.; Rhee, S.Y.; Libiran, P.; Trevino, C.; Fessel, W.J.; Shafer, R.W. HIV-1 integrase sequence variability in antiretroviral naïve patients and in triple-class experienced patients subsequently treated with raltegravir. AIDS Res. Hum. Retroviruses 2010, 26, 1323–1326. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.E.; Pillay, D. Analysis of natural sequence variation and covariation in human immunodeficiency virus type 1 integrase. J. Virol. 2008, 82, 9228–9235. [Google Scholar] [CrossRef] [Green Version]
- Bukovsky, A.; Göttlinger, H. Lack of integrase can markedly affect human immunodeficiency virus type 1 particle production in the presence of an active viral protease. J. Virol. 1996, 70, 6820–6825. [Google Scholar] [CrossRef] [Green Version]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS ONE 2013, 8, e74163. [Google Scholar] [CrossRef] [PubMed]
- Hoyte, A.C.; Jamin, A.V.; Koneru, P.C.; Kobe, M.J.; Larue, R.C.; Fuchs, J.R.; Engelman, A.N.; Kvaratskhelia, M. Resistance to pyridine-based inhibitor KF116 reveals an unexpected role of integrase in HIV-1 Gag-Pol polyprotein proteolytic processing. J. Biol. Chem. 2017, 292, 19814–19825. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IN:50 * | RH:79 * | IN:17 * | Sequence #† | Patient #‡ | T.CD4 (Cells/μL) ** | VL (Copies/mL) |
|---|---|---|---|---|---|---|
| M50M *** | N79N | S17S | 139 | 55 | 3–227 | 874–2,121,990 |
| M50I | N79N | S17S | 14 | 8 | 222–705 | 1207–201,353 |
| M50I | N79N | S17N | 13 | 3 | 174–940 | 2024–741,044 |
| M50I | N79S | S79S | 36 | 15 | 110–734 | 1529–1,009,970 |
| M50I | N79S | S17N | 23 | 7 | 18–589 | 4677–977,945 |
| M50I | N79S | S17C | 2 | 1 | 46–52 | 786,337–834,397 |
| M50I | N79S | S17T | 2 | 1 | 452–476 | 3337–4893 |
| IN:50 * | |||||||
|---|---|---|---|---|---|---|---|
| M50I ** | M50L | M50M | M50R | M50T | Samples/Patients # | ||
| IN:151 * | V151I | 0 ## | 0 | 3/2 | 0 | 11/2 | 14/4 (2%) ☨ |
| V151V | 90/35 | 5/2 | 404/161 | 2/2 | 14/5 | 515/205 (98%) | |
| Sample # | 90/35 | 5/2 | 407/163 | 2/2 | 25/7 | 529/209 (100%) | |
| Subtype | M50I | V151I | M50I/V151I | Total ** |
|---|---|---|---|---|
| A | 10.89% | 0.00% | 0.00% | 1194 |
| B | 15.27% | 3.22% | 0.43% | 6335 |
| C | 47.69% | 0.31% | 0.10% | 2871 |
| D | 1.54% | 0.77% | 0.00% | 260 |
| F | 10.42% | 1.39% | 0.00% | 144 |
| G | 15.97% | 0.69% | 0.00% | 144 |
| H | 14.29% | 0.00% | 0.00% | 14 |
| J | 10.00% | 0.00% | 0.00% | 10 |
| K | 66.67% | 0.00% | 0.00% | 3 |
| L | 33.33% | 0.00% | 0.00% | 3 |
| U | 22.50% | 0.00% | 0.00% | 40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Hao, M.; Khan, M.A.; Rehman, M.T.; Highbarger, H.C.; Chen, Q.; Goswami, S.; Sherman, B.T.; Rehm, C.A.; Dewar, R.L.; et al. A Combination of M50I and V151I Polymorphic Mutations in HIV-1 Subtype B Integrase Results in Defects in Autoprocessing. Viruses 2021, 13, 2331. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112331
Yang J, Hao M, Khan MA, Rehman MT, Highbarger HC, Chen Q, Goswami S, Sherman BT, Rehm CA, Dewar RL, et al. A Combination of M50I and V151I Polymorphic Mutations in HIV-1 Subtype B Integrase Results in Defects in Autoprocessing. Viruses. 2021; 13(11):2331. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112331
Chicago/Turabian StyleYang, Jun, Ming Hao, Muhammad A. Khan, Muhammad T. Rehman, Helene C. Highbarger, Qian Chen, Suranjana Goswami, Brad T. Sherman, Catherine A. Rehm, Robin L. Dewar, and et al. 2021. "A Combination of M50I and V151I Polymorphic Mutations in HIV-1 Subtype B Integrase Results in Defects in Autoprocessing" Viruses 13, no. 11: 2331. https://0-doi-org.brum.beds.ac.uk/10.3390/v13112331