
Molecular Characterization of Small Ruminant Lentiviruses in Polish Mixed Flocks Supports Evidence of Cross Species Transmission, Dual Infection, a Recombination Event, and Reveals the Existence of New Subtypes within Group A


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Samples
2.2. PCR Technique
2.3. DNA Sequencing and Analysis
2.4. Analysis of Recombination
3. Results
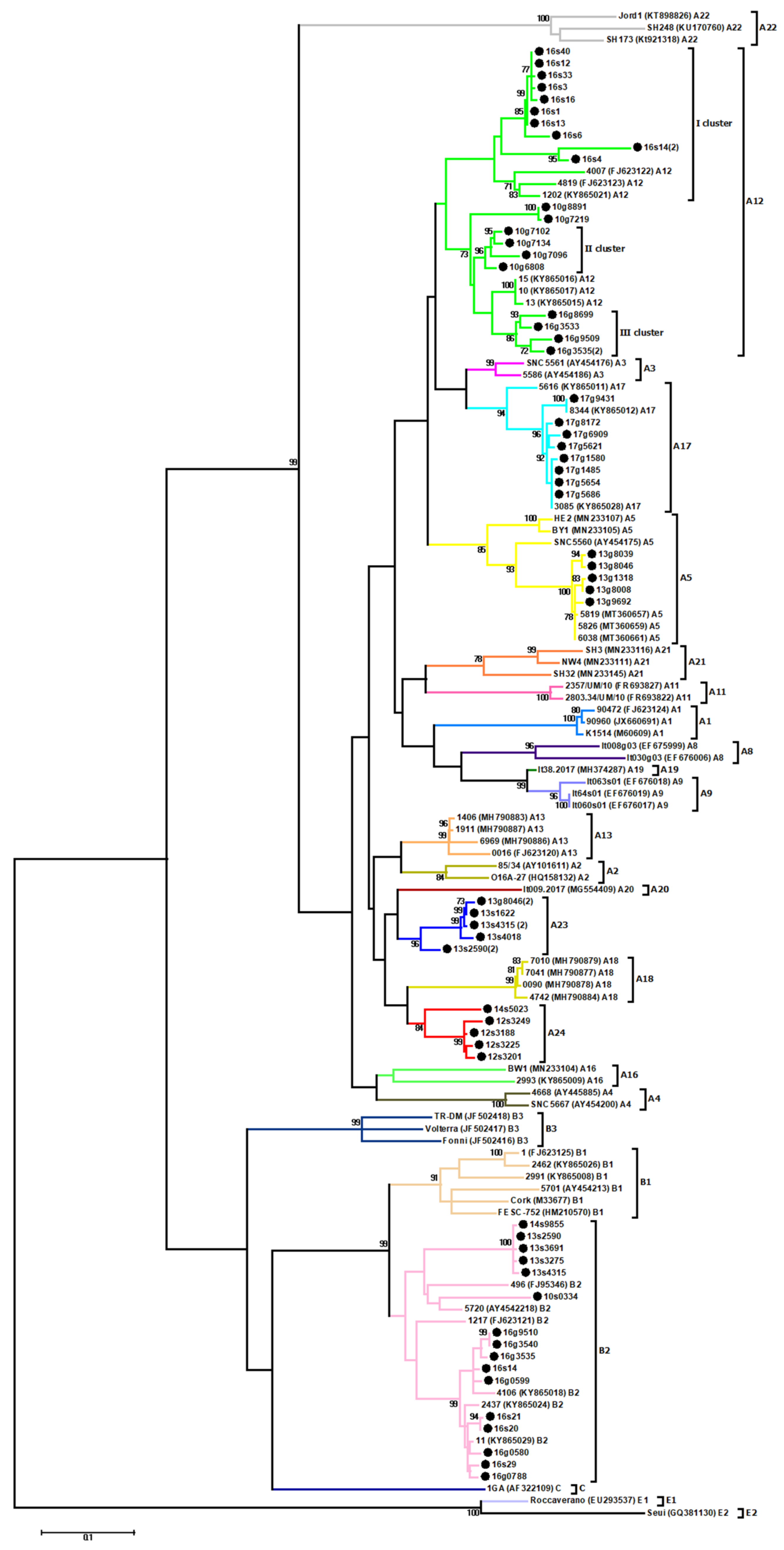
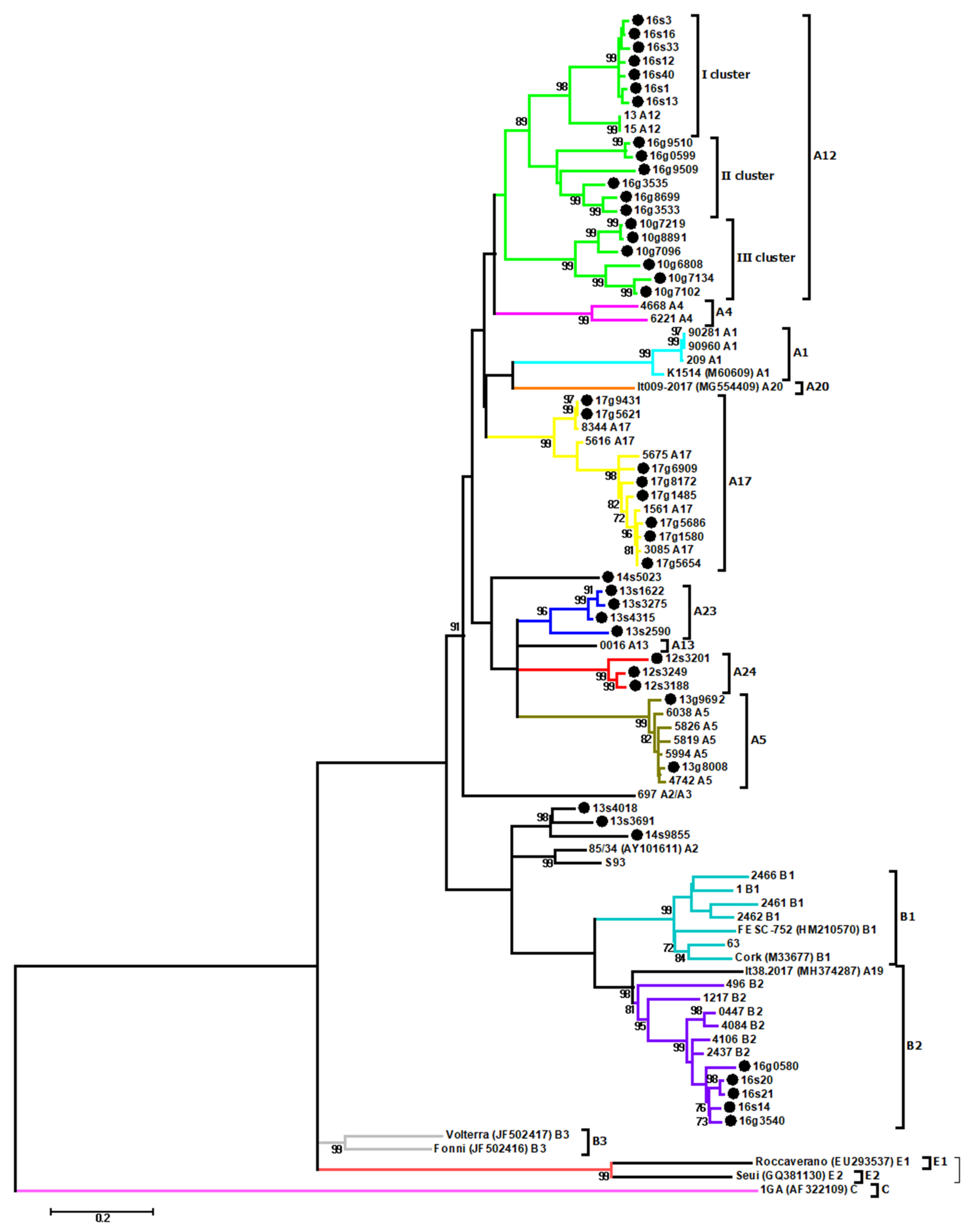
3.1. Phylogenetic Analysis
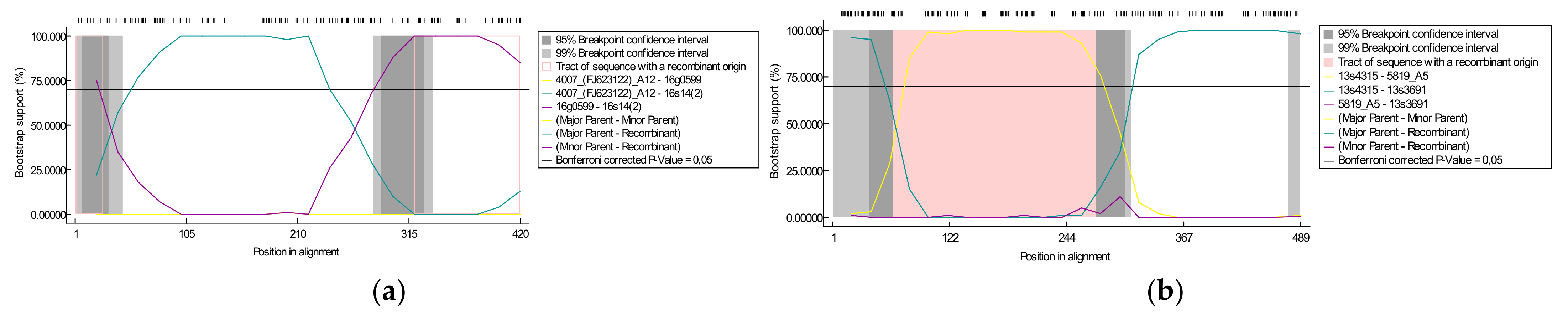
3.2. Identification of Putative Recombination
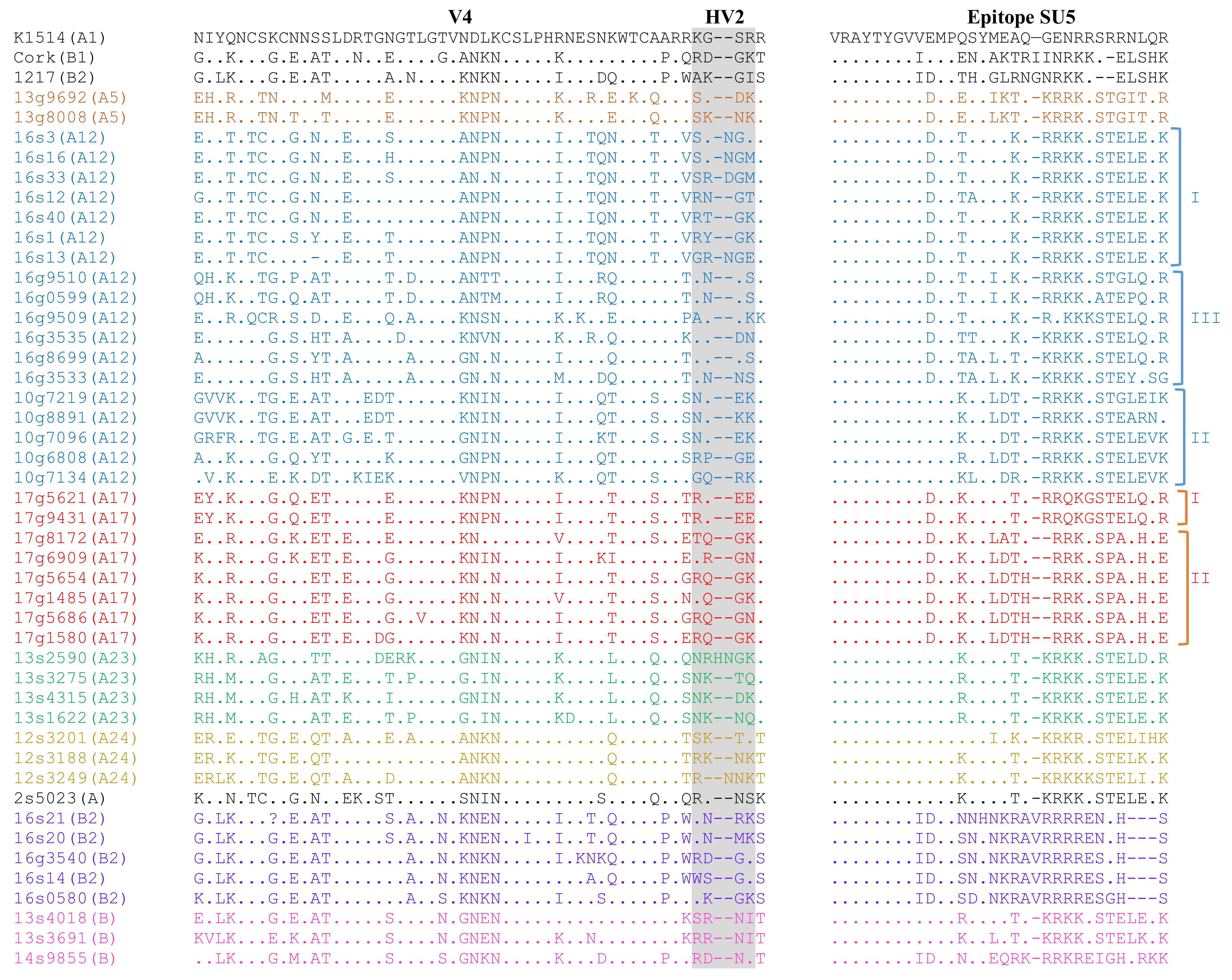
3.3. Analysis of Immunodominant Regions
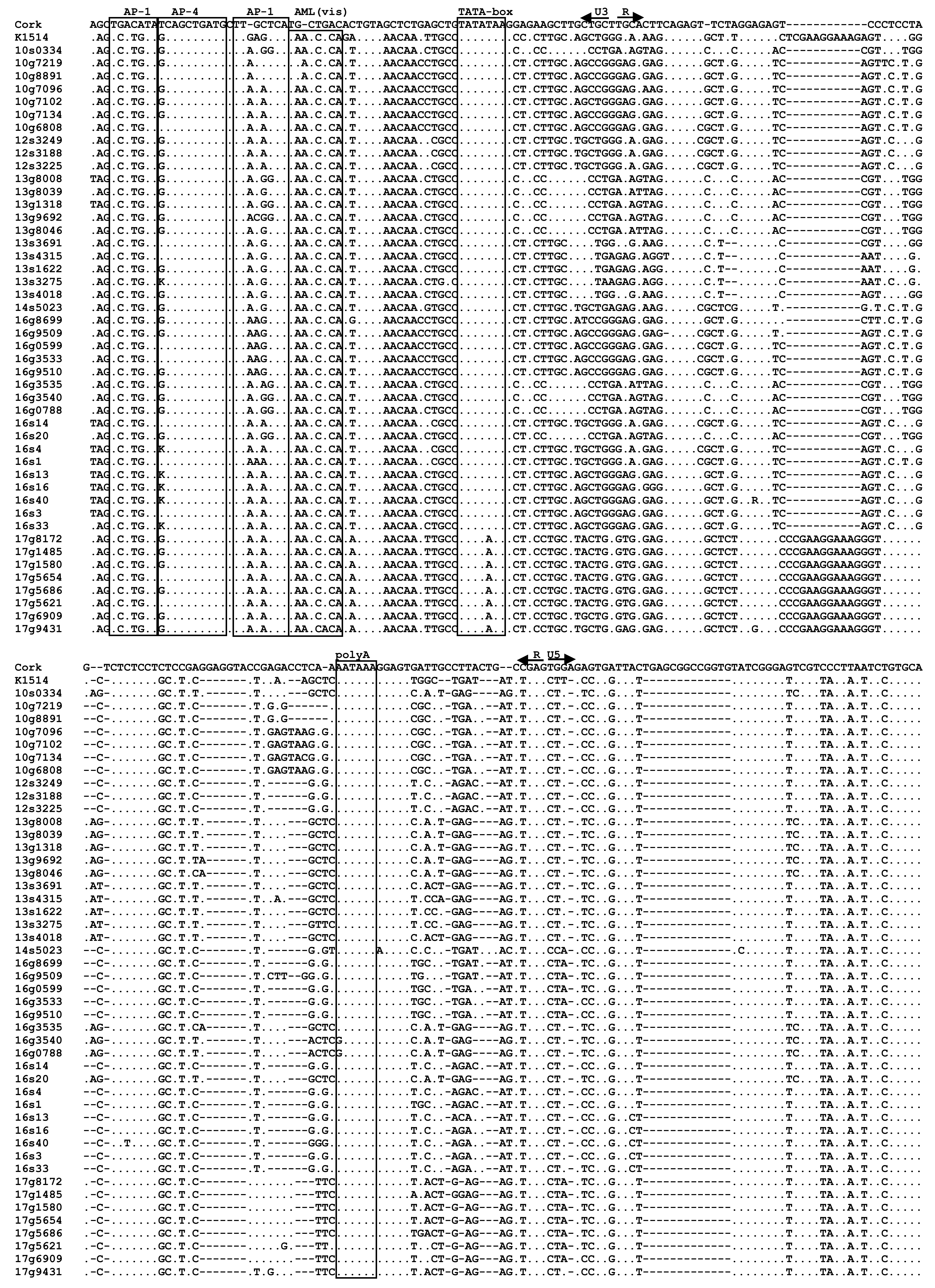
3.4. Analysis of LTR Sequences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Minardi da Cruz, J.C.; Singh, D.K.; Lamara, A.; Chebloune, Y. Small ruminant lentiviruses (SRLVs) break the species barrier to acquire new host range. Viruses 2013, 5, 1867–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, O.; Clements, J.E.; Strandberg, J.D.; Cork, L.C.; Griffin, D.E. Biological characterization of the virus causing leukoencephalitis and arthritis in goats. J. Gen. Virol. 1980, 50, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Peterhans, E.; Greenland, T.; Badiola, J.; Harkiss, G.; Bertoni, G.; Amorena, B.; Eliaszewicz, M.; Juste, R.; Kraßnig, R.; Lafont, J.-P.; et al. Routes of transmission and consequences of small ruminant lentiviruses (SRLVs) infection and eradication schemes. Vet. Res. 2004, 35, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Minguijón, E.; Reina, R.; Pérez, M.; Polledo, L.; Villoria, M.; Ramírez, H.; Leginagoikoa, I.; Badiola, J.J.; García-Marín, J.F.; de Andrés, D.; et al. Small ruminant lentivirus infections and diseases. Vet. Microbiol. 2015, 181, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Villet, S.; Bouzar, B.A.; Morin, T.; Verdier, G.; Legras, C.; Chebloune, Y. Maedi-visna virus and caprine arthritis encephalitis virus genomes encode a Vpr-like but no Tat protein. J. Virol. 2003, 77, 9632–9638. [Google Scholar] [CrossRef] [Green Version]
- Pepin, M.; Vitu, C.; Russo, P.; Mornex, J.F.; Peterhans, E. Meadi-Visna virus infection in sheep: A review. Vet. Res. 1998, 29, 341–367. [Google Scholar]
- Saltarelli, M.; Querat, G.; Konings, D.A.M.; Vigne, R.; Clements, J.E. Nucleotide sequence and transcriptional analysis of molecular clones of CAEV which generate infectious virus. Virology 1990, 179, 347–364. [Google Scholar] [CrossRef]
- Gomez-Lucia, E.; Barquero, N.; Domenech, A. Maedi-Visna virus: Current perspectives. Vet. Med. 2018, 9, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molaee, V.; Bazzucchi, M.; De Mia, G.M.; Otarod, V.; Abdollahi, D.; Rosati, S.; Lühken, G. Phylogenetic analysis of small ruminant lentiviruses in Germany and Iran suggests their expansion with domestic sheep. Sci. Rep. 2020, 10, 2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, R.; Adjadj, N.R.; De Regge, N. Phylogenetic Analysis of Belgian Small Ruminant Lentiviruses Supports Cross Species Virus Transmission and Identifies New Subtype B5 Strains. Pathogens 2020, 9, 183. [Google Scholar] [CrossRef] [Green Version]
- Reina, R.; Bertolotti, L.; Giudici, S.D.; Puggioni, G.; Ponti, N.; Profiti, M.; Patta, C.; Rosati, S. Small Ruminant Lentivirus Genotype E Is Widespread in Sarda Goat. Vet. Microbiol. 2010, 144, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reina, R.; Mora, M.I.; Glaria, I.; García, I.; Solano, C.; Luján, L.; Badiola, J.J.; Contreras, A.; Berriatua, E.; Juste, R.; et al. Molecular characterization and phylogenetic study of Maedi Visna and Caprine Arthritis Encephalitis viral sequences in sheep and goats from Spain. Virus Res. 2006, 121, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Colitti, B.; Coradduzza, E.; Puggioni, G.; Capucchio, M.T.; Reina, R.; Bertolotti, L.; Rosati, S. A new approach for Small Ruminant Lentivirus full genome characterization revealed the circulation of divergent strains. PLoS ONE 2019, 14, e0212585. [Google Scholar] [CrossRef] [PubMed]
- Gjerset, B.; Storset, A.K.; Rimstad, E. Genetic diversity of small-ruminant lentiviruses: Characterization of Norwegian isolates of Caprine arthritis encephalitis virus. J. Gen. Virol. 2006, 87, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Gjerset, B.; Rimstad, E.; Teige, J.; Soetaert, K.; Jonassen, C.M. Impact of natural sheep-goat transmission on detection and control of small ruminant lentivirus group C infections. Vet. Microbiol. 2009, 135, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Pisoni, G.; Quasso, A.; Moroni, P. Phylogenetic analysis of small-ruminant lentivirus subtype B1 in mixed flocks: Evidence for natural transmission from goats to sheep. Virology 2005, 339, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Leroux, C.; Cruz, J.C.; Mornex, J.F. SRLVs: A genetic continuum of lentiviral species in sheep and goats with cumulative evidence of cross species transmission. Curr. HIV Res. 2010, 8, 94–100. [Google Scholar]
- Fras, M.; Leboeuf, A.; Labrie, F.M.; Laurin, M.A.; Singh Sohal, J.; L’Homme, Y. Phylogenetic analysis of small ruminant lentiviruses in mixed flocks: Multiple evidence of dual infection and natural transmission of types A2 and B1 between sheep and goats. Infect. Genet. Evol. 2013, 19, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Grego, E.; Bertolotti, L.; Quasso, A.; Profiti, M.; Lacerenza, D.; Muz, D.; Rosati, S. Genetic characterization of small ruminant lentivirus in Italian mixed flocks: Evidence for a novel genotype circulating in a local goat population. J. Gen. Virol. 2007, 88, 3423–3427. [Google Scholar] [CrossRef] [PubMed]
- Santry, L.A.; de Jong, J.; Gold, A.C.; Walsh, S.R.; Menzies, P.I.; Wootton, S.K. Genetic characterization of small ruminant lentiviruses circulating in naturally infected sheep and goats in Ontario, Canada. Virus Res. 2013, 175, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Olech, M.; Osiński, Z.; Kuźmak, J. Bayesian estimation of seroprevalence of small ruminant lentiviruses in sheep from Poland. Prev. Veter Med. 2017, 147, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Kaba, J.; Czopowicz, M.; Ganter, M.; Nowicki, M.; Witkowski, L.; Nowicka, D.; Szaluś-Jordanow, O. Risk factors associated with seropositivity to small ruminant lentiviruses in goat herds. Res. Vet. Sci. 2013, 94, 225–227. [Google Scholar] [CrossRef]
- Olech, M.; Rachid, A.; Croisé, B.; Kuźmak, J.; Valas, S. Genetic and antigenic characterization of small ruminant lentiviruses circulating in Poland. Virus Res. 2012, 163, 528–536. [Google Scholar] [CrossRef]
- Olech, M.; Valas, S.; Kuźmak, J. Epidemiological survey in single-species flocks from Poland reveals expanded genetic and antigenic diversity of small ruminant lentiviruses. PLoS ONE 2018, 13, e193892. [Google Scholar] [CrossRef] [Green Version]
- Olech, M.; Murawski, M.; Kuźmak, J. Molecular analysis of small-ruminant lentiviruses in Polish flocks reveals the existence of a novel subtype in sheep. Arch. Virol. 2019, 164, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olech, M.; Kuźmak, J. Molecular Characterization of Small Ruminant Lentiviruses of Subtype A5 Detected in Naturally Infected but Clinically Healthy Goats of Carpathian Breed. Pathogens 2020, 9, 992. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Tiley, L.; McConnell, I.; Blacklaws, B. Infection of dendritic cells by the Maedi-Visna lentivirus. J. Virol. 2000, 74, 10096–10103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordasini, F.; Vogt, H.R.; Zahno, M.L.; Maeschli, A.; Nenci, C.; Zanoni, R.; Peterhans, E.; Bertoni, G. Analysis of the antibody response to an immunodominant epitope of the envelope glycoprotein of a lentivirus and its diagnostic potential. J. Clin. Microbiol. 2006, 44, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Korber, B.; Rodrigo, A.G.; Learn, G.H. HIV Signature and Sequence Variation Analysis, Computational Analysis of HIV Molecular Sequences; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; pp. 55–72. [Google Scholar]
- Zhang, M.; Gaschen, B.; Blay, W.; Foley, B.; Haigwood, N.; Kuiken, C.; Korber, B. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 2004, 14, 1229–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.S.; Joshi, S.B. Reusable software concepts applied to the development of fms control software. Int. J. Comput. Integr. Manuf. 1992, 5, 182–196. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in rebombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, C.; Böni, J.; Huder, J.B.; Vogt, H.-R.; Mühlherr, J.; Zanoni, R.; Miserez, R.; Lutz, H.; Schüpbach, J. Phylogenetic analysis and reclassification of caprine and ovine lentiviruses based on 104 new isolates: Evidence for regular sheep-to-goat transmission and worldwide propagation through livestock trade. Virology 2004, 319, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Muz, D.; Oğuzoğlu, T.C.; Rosati, S.; Reina, R.; Bertolotti, L.; Burgu, I. First molecular characterization of visna/maedi viruses from naturally infected sheep in Turkey. Arch. Virol. 2013, 158, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Kokawa, S.; Oba, M.; Hirata, T.; Tamaki, S.; Omura, M.; Tsuchiaka, S.; Nagai, M.; Omatsu, T.; Mizutani, T. Molecular characteristics and prevalence of small ruminant lentiviruses in goats in Japan. Arch Virol. 2017, 162, 3007–3015. [Google Scholar] [CrossRef]
- Smyth, R.P.; Davenport, M.P.; Mak, J. The origin of genetic diversity in HIV-1. Virus Res. 2012, 169, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Pecon-Slattery, J.; Troyer, J.L.; Johnson, W.E.; O’Brien, S.J. Evolution of feline immunodeficiency virus in Felidae: Implications for human health and wildlife ecology. Vet. Immunol. Immunopathol. 2008, 123, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Pisoni, G.; Bertoni, G.; Puricelli, M.; Maccalli, M.; Moroni, P. Demonstration of coinfection with and recombination by caprine arthritis-encephalitis virus and maedi-visna virus in naturally infected goats. J. Virol. 2007, 81, 4948–4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, H.; Reina, R.; Bertolotti, L.; Cenoz, A.; Hernández, M.M.; San Román, B.; Glaria, I.; de Andrés, X.; Crespo, H.; Jáuregui, P.; et al. Study of compartmentalization in the visna clinical form of small ruminant lentivirus infection in sheep. BMC Vet. Res. 2012, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisoni, G.; Bertoni, G.; Boettcher, P.; Ponti, W.; Moroni, P. Phylogenetic analysis of the gag region encoding the matrix protein of small ruminant lentiviruses: Comparative analysis and molecular epidemiological applications. Virus Res. 2006, 116, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, L.; Mazzei, M.; Puggioni, G.; Carrozza, M.L.; Dei Giudici, S.; Muz, D.; Juganaru, M.; Patta, C.; Tolari, F.; Rosati, S. Characterization of new small ruminant lentivirus subtype B3 suggests animal trade within the Mediterranean Basin. J. Gen. Virol. 2011, 92, 1923–1929. [Google Scholar] [CrossRef]
- Pisoni, G.; Bertoni, G.; Manarolla, G.; Vogt, H.R.; Scaccabarozzi, L.; Locatelli, C.; Moroni, P. Genetic analysis of small ruminant lentiviruses following lactogenic transmission. Virology 2010, 407, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Dickey, A.M.; Smith, T.P.L.; Clawson, M.L.; Heaton, M.P.; Workman, A.M. Classification of small ruminant lentivirus subtype A2, subgroups 1 and 2 based on whole genome comparisons and complex recombination patterns [version 2; peer review: 1 approved, 1 approved with reservations]. F1000Research 2021, 9, 1449. [Google Scholar] [CrossRef]
- Glaria, I.; Reina, R.; Crespo, H.; de Andres, X.; Ramirez, H.; Biescas, E.; Perez, M.M.; Badiola, J.; Lujan, L.; Amorena, B.; et al. Phylogenetic analysis of SRLV sequences from an arthritic sheep outbreak demonstrates the introduction of CAEV-like viruses among Spanish sheep. Vet. Microbiol. 2009, 138, 156–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, K.M.; Bildfell, R.J.; Anderson, M.L.; Cebra, C.K.; Valentine, B.A. Fatal Caprine arthritis encephalitis virus-like infection in 4 Rocky Mountain goats (Oreamnos americanus). J. Vet. Diagn. Invest. 2012, 24, 392–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, T.; Guiguen, F.; Bouzar, B.A.; Villet, S.; Greenland, T.; Grezel, D.; Gounel, F.; Gallay, K.; Garnier, C.; Durand, J.; et al. Clearance of a productive lentivirus infection in calves experimentally inoculated with caprine arthritis-encephalitis virus. J. Virol. 2003, 77, 6430–6437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosati, S.; Mannelli, A.; Merlo, T.; Ponti, N. Characterization of the immunodominant cross-reacting epitope of visna maedi virus and caprine arthritis-encephalitis virus capsid antigen. Virus Res. 1999, 61, 177–183. [Google Scholar] [CrossRef]
- Kalogianni, A.I.; Stavropoulos, I.; Chaintoutis, S.C.; Bossis, I.; Gelasakis, A.I. Serological, Molecular and Culture-Based Diagnosis of Lentiviral Infections in Small Ruminants. Viruses 2021, 13, 1711. [Google Scholar] [CrossRef]
- Grego, E.; Profiti, M.; Giammarioli, M.; Giannino, L.; Rutili, D.; Woodall, C.; Rosati, S. Genetic heterogeneity of small ruminant lentiviruses involves immunodominant epitope of capsid antigen and affects sensitivity of single-strain-based immunoassay. Clin. Diagn. Lab. Immunol. 2002, 9, 828–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosati, S.; Profiti, M.; Grego, E.; Carrozza, M.L.; Mazzei, M.; Bandecchi, P. Antigenic variability of ovine lentivirus isolated in Italy. Vet. Res. Commun. 2004, 28, 319–322. [Google Scholar] [CrossRef]
- Purdy, J.B.; Freeman, A.F.; Martin, S.C.; Ryder, C.; Elliott-DeSorbo, D.K.; Zeichner, S.; Hazra, R. Virologic response using directly observed therapy in adolescents with HIV: An adherence tool. J. Assoc. Nurses AIDS Care 2008, 19, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.H.; Chang, Y.F.; Wang, C.T. Mutations in the alpha-helix directly C-terminal to the major homology region of human immunodeficiency virus type 1 capsid protein disrupt Gag multimerization and markedly impair virus particle production. J. Biomed. Sci. 2006, 13, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Valas, S.; Benoit, C.; Baudry, C.; Perrin, G.; Mamoun, R.Z. Variability and immunogenicity of caprine arthritis encephalitis virus surface glycoprotein. J. Virol. 2000, 74, 6178–6185. [Google Scholar] [CrossRef] [Green Version]
- Hötzel, I.; Kumpula-McWhirter, N.; Cheevers, W.P. Rapid evolution of two discrete regions of the caprine arthritis-encephalitis virus envelope surface glycoprotein during persistent infection. Virus Res. 2002, 84, 17–25. [Google Scholar] [CrossRef]
- Skraban, R.; Matthíasdóttir, S.; Torsteinsdóttir, S.; Agnarsdóttir, G.; Gudmundsson, B.; Georgsson, G.; Meloen, R.H.; Andrésson, Ó.S.; Staskus, K.A.; Thormar, H.; et al. Naturally occurring mutations within 39 amino acids in the envelope glycoprotein of maedi-visna virus alter the neutralization phenotype. J. Virol. 1999, 73, 8064–8072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, H.; Reina, R.; Amorena, B.; de Andrés, D.; Martínez, H.A. Small ruminant lentiviruses: Genetic variability, tropism and diagnosis. Viruses 2013, 5, 1175–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González Méndez, A.S.; Cerón Téllez, F.; Tórtora Pérez, J.L.; Martínez Rodríguez, H.A.; García Flores, M.M.; Ramírez Álvarez, H. Signature patterns in region V4 of small ruminant lentivirus surface protein in sheep and goats. Virus Res. 2020, 280, 197900. [Google Scholar] [CrossRef] [PubMed]
- Zahno, M.L.; Bertoni, G. An Immunodominant Region of the Envelope Glycoprotein of Small Ruminant Lentiviruses May Function as Decoy Antigen. Viruses 2018, 10, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Nie, J.; Prochnow, C.; Truong, C.; Jia, Z.; Wang, S.; Chen, X.S.; Wang, Y. A systematic study of the N-glycosylation sites of HIV-1 envelope protein on infectivity and antibody-mediated neutralization. Retrovirology 2013, 10, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, A.F.; Lewis, F.I.; Pond, S.L.; Frost, S.D. Evolutionary interactions between N-linked glycosylation sites in the HIV-1 envelope. PLoS Comput. Biol. 2007, 3, e11. [Google Scholar] [CrossRef]
- Gayo, E.; Cuteri, V.; Polledo, L.; Rossi, G.; García Marín, J.F.; Preziuso, S. Genetic Characterization and Phylogenetic Analysis of Small Ruminant Lentiviruses Detected in Spanish Assaf Sheep with Different Mammary Lesions. Viruses 2018, 10, 315. [Google Scholar] [CrossRef] [Green Version]
- Angelopoulou, K.; Brellou, G.D.; Greenland, T.; Vlemmas, I. A novel deletion in the LTR region of a Greek small ruminant lentivirus may be associated with low pathogenicity. Virus Res. 2006, 118, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lucia, E.; Rowe, J.; Collar, C.; Murphy, B. Diversity of caprine arthritis-encephalitis virus promoters isolated from goat milk and passaged in vitro. Vet. J. 2013, 196, 431–438. [Google Scholar] [CrossRef]
- Mendiola, W.P.S.; Tórtora, J.L.; Martínez, H.A.; García, M.M.; Cuevas-Romero, S.; Cerriteño, J.L.; Ramírez, H. Genotyping Based on the LTR Region of Small Ruminant Lentiviruses from Naturally Infected Sheep and Goats from Mexico. Biomed. Res. Int. 2019, 2019, 4279573. [Google Scholar] [CrossRef]
- van Opijnen, T.; Kamoschinski, J.; Jeeninga, R.E.; Berkhout, B. The human immunodeficiency virus type 1 promoter contains a CATA box instead of a TATA box for optimal transcription and replication. J. Virol. 2004, 78, 6883–6890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller-Jensen, K.; Skupsky, R.; Shah, P.S.; Arkin, A.P.; Schaffer, D.V. Genetic selection for context-dependent stochastic phenotypes: Sp1 and TATA mutations increase phenotypic noise in HIV-1 gene expression. PLoS Comput. Biol. 2013, 9, e1003135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsson, T.; Hreggvidsdóttir, H.S.; Agnarsdóttir, G.; Matthíasdóttir, S.; Ogmundsdóttir, M.H.; Jónsson, S.R.; Georgsson, G.; Ingvarsson, S.; Andrésson, O.S.; Andrésdóttir, V. Duplicated Sequence Motif in the Long Terminal Repeat of Maedi-Visna Virus Extends Cell Tropism and Is Associated with Neurovirulence. J. Virol. 2007, 81, 4052–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pair | Primers | Sequences (5′-3′) | Orientation | PCR | Product Length (bp) |
|---|---|---|---|---|---|
| LTR | |||||
| A | LTREFW | ACTGTCAGGRCAGAGAACARATGCC | F | 1 | 407 |
| LTRERV | CTCTCTTACCTTACTTCAGG | R | 1 | ||
| B | LTRIFW | AAGTCATGTAKCAGCTGATGCTT | F | 2 | 213 |
| LTRIRV | TTGCACGGAATTAGTAACG | R | 2 | ||
| Env | |||||
| C | 423 | GGRGCAGARATMATHCCWGAARVYHTGA | F | 1 | 1205 |
| 564 | GCYAYATGCTGIACCATGGCATA | R | 1 | ||
| D | 423 | GGRGCAGARATMATHCCWGAARVYHTGA | F | 2 | 1076 |
| 425 | CCTGCRGCAGCYAYTATHGCCAT | R | 2 | ||
| E | 563 | GAYATGRYRGARCAYATGAC | F | 2 | 818 |
| 564 | GCYAYATGCTGIACCATGGCATA | R | 2 | ||
| F | 563 | GAYATGRYRGARCAYATGAC | F | 2 | 689 |
| 425 | CCTGCRGCAGCYAYTATHGCCAT | R | 2 | ||
| G | 567 | GGIACIAAIACWAATTGGAC | F | 2 | 608 |
| 564 | GCYAYATGCTGIACCATGGCATA | R | 2 | ||
| Gag | |||||
| H | GAGf1 | TGGTGARKCTAGMTAGAGACATGG | F | 1 | 1350 |
| P15 | GTTATTCCATAGGAGGAGCGGACGGCACCA | R | 1 | ||
| I | CAGAG5 | GCRGGRGGGAAGRAGYTGGAA | F | 2 | 625 |
| CAGAG3 | ACATGCTTGCATTTTTTYTTCTAC | R | 2 |
| GAG | ENV | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample No. | Flock | Region | Host | Strain | GenBank Accession Number | Proposed Subtype | GenBank Accession Number | Proposed Subtype |
| 1 | 10 | Wielkopolskie | goat | 7134 | OL348032 | A12 | OL436271 | A12 |
| 2 | goat | 7102 | OL348031 | A12 | OL436303 | A12 | ||
| 3 | goat | 6808 | OL348029 | A12 | OL436270 | A12 | ||
| 4 | goat | 7219 | OL348023 | A12 | OL436269 | A12 | ||
| 5 | goat | 7096 | OL348030 | A12 | OL436268 | A12 | ||
| 6 | goat | 8891 | OL348024 | A12 | OL436297 | A12 | ||
| 7 | sheep | 0334 | OL348005 | B2 | N/A | N/A | ||
| 8 | 12 | Podkarpackie | sheep | 3225 | OL348051 | A24 | N/A | N/A |
| 9 | sheep | 3201 | OL348052 | A24 | OL436300 | A24 | ||
| 10 | sheep | 3188 | OL348050 | A24 | OL436296 | A24 | ||
| 11 | sheep | 3249 | OL348049 | A24 | OL436302 | A24 | ||
| 12 | 13 | Podkarpackie | goat | 1318 | OL348020 | A5 | N/A | N/A |
| 13 | goat | 8008 | OL348021 | A5 | OL436267 | A5 | ||
| 14 | goat | 9692 | OL348019 | A5 | OL436266 | A5 | ||
| 15 | goat | 8039 | OL348017 | A5 | N/A | N/A | ||
| 16 | goat | 8046 | OL348018, OL348056 | A5/A23 | N/A | N/A | ||
| 17 | sheep | 4315 | OL348001, OL348058 | A23/B2 | OL436285 | A23 | ||
| 18 | sheep | 1622 | OL348057 | A23 | OL436287 | A23 | ||
| 19 | sheep | 4018 | OL348055 | A23 | OL436260 | B | ||
| 20 | sheep | 2590 | OL348004, OL348054 | A23/B2 | OL436284 | A23 | ||
| 21 | sheep | 3691 | OL348002 | B2 | OL436261 | B | ||
| 22 | sheep | 3275 | OL348003 | B2 | OL436286 | A23 | ||
| 23 | 14 | Podkarpackie | sheep | 9855 | OL348000 | B2 | OL436298 | B |
| 24 | sheep | 5023 | OL348053 | A24 | OL436272 | A | ||
| 25 | 16 | Lubelskie | sheep | 40 | OL348037 | A12 | OL436276 | A12 |
| 26 | sheep | 33 | OL348035 | A12 | OL436299 | A12 | ||
| 27 | sheep | 3 | OL348036 | A12 | OL436259 | A12 | ||
| 28 | sheep | 12 | OL348038 | A12 | OL436275 | A12 | ||
| 29 | sheep | 16 | OL348039 | A12 | OL436277 | A12 | ||
| 30 | sheep | 1 | OL348034 | A12 | OL436273 | A12 | ||
| 31 | sheep | 13 | OL348040 | A12 | OL436274 | A12 | ||
| 32 | sheep | 6 | OL348033 | A12 | N/A | N/A | ||
| 33 | sheep | 4 | OL348022 | A12 | N/A | N/A | ||
| 34 | sheep | 14 | OL348006, OL348016 | A12/B2 | OL436264 | B2 | ||
| 35 | sheep | 21 | OL348014 | B2 | OL436301 | B2 | ||
| 36 | sheep | 20 | OL348015 | B2 | OL436263 | B2 | ||
| 37 | sheep | 29 | OL348011 | B2 | N/A | N/A | ||
| 38 | goat | 8699 | OL348025 | A12 | OL436282 | A12 | ||
| 39 | goat | 3533 | OL348026 | A12 | OL436283 | A12 | ||
| 40 | goat | 3535 | OL348010, OL348028 | A12/B2 | OL436281 | A12 | ||
| 41 | goat | 9509 | OL348027 | A12 | OL436280 | A12 | ||
| 42 | goat | 9510 | OL348008 | B2 | OL436278 | A12 | ||
| 43 | goat | 3540 | OL348009 | B2 | OL436265 | B2 | ||
| 44 | goat | 0599 | OL348007 | B2 | OL436279 | A | ||
| 45 | goat | 0788 | OL348012 | B2 | N/A | N/A | ||
| 46 | goat | 0580 | OL348013 | B2 | OL436262 | B2 | ||
| 47 | 17 | Mazowieckie | goat | 1485 | OL348044 | A17 | OL436290 | A17 |
| 48 | goat | 5654 | OL348045 | A17 | OL436292 | A17 | ||
| 49 | goat | 5686 | OL348046 | A17 | OL436291 | A17 | ||
| 50 | goat | 1580 | OL348043 | A17 | OL436293 | A17 | ||
| 51 | goat | 5621 | OL348048 | A17 | OL436295 | A17 | ||
| 52 | goat | 6909 | OL348047 | A17 | OL436288 | A17 | ||
| 53 | goat | 8172 | OL348042 | A17 | OL436289 | A17 | ||
| 54 | goat | 9431 | OL348041 | A17 | OL436294 | A17 | ||
| A1 | A2 | A3 | A4 | A5 | A8 | A9 | A11 | A12 | A13 | A16 | A17 | A18 | A19 | A20 | A21 | A22 | A23 | A24 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A2 | 18.6 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A3 | 17.9 | 14.5 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A4 | 18.5 | 19.0 | 19.1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A5 | 19.2 | 17.4 | 15.0 | 17.8 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A8 | 19.4 | 19.4 | 18.0 | 19.7 | 19.8 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A9 | 18.7 | 17.1 | 14.6 | 18.8 | 16.7 | 16.9 | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A11 | 19.0 | 17.3 | 17.6 | 22.2 | 19.2 | 19.0 | 16.4 | - | - | - | - | - | - | - | - | - | - | - | - |
| A12 | 20.4 | 15.0 | 13.0 | 19.5 | 15.9 | 19.0 | 18.3 | 16.7 | - | - | - | - | - | - | - | - | - | - | - |
| A13 | 18.3 | 11.6 | 14.4 | 17.0 | 16.0 | 19.6 | 18.8 | 16.2 | 15.1 | - | - | - | - | - | - | - | - | - | - |
| A16 | 21.2 | 17.7 | 20.6 | 18.3 | 18.7 | 22.3 | 22.4 | 21.3 | 19.3 | 17.7 | - | - | - | - | - | - | - | - | - |
| A17 | 16.6 | 13.6 | 10.9 | 17.0 | 13.8 | 18.0 | 13.7 | 18.1 | 13.3 | 15.2 | 17.4 | - | - | - | - | - | - | - | - |
| A18 | 21.6 | 13.5 | 17.1 | 20.2 | 18.9 | 22.6 | 19.9 | 20.1 | 16.7 | 14.0 | 18.7 | 16.1 | - | - | - | - | - | - | - |
| A19 | 18.6 | 15.4 | 13.4 | 17.2 | 14.9 | 16.2 | 5.1 | 15.3 | 16.7 | 16.5 | 20.1 | 12.6 | 17.8 | - | - | - | - | - | - |
| A20 | 21.9 | 16.3 | 17.2 | 19.3 | 16.9 | 20.6 | 18.9 | 17.8 | 17.1 | 16.9 | 18.6 | 18.0 | 18.5 | 16.8 | - | - | - | - | - |
| A21 | 18.5 | 16.3 | 16.1 | 21.9 | 17.0 | 21.5 | 19.0 | 17.0 | 16.5 | 15.4 | 19.7 | 17.4 | 17.2 | 18.2 | 19.3 | - | - | - | - |
| A22 | 28.0 | 23.9 | 22.5 | 27.3 | 22.1 | 27.1 | 25.7 | 26.6 | 24.9 | 25.8 | 27.5 | 23.6 | 23.6 | 25.3 | 23.6 | 23.9 | - | - | - |
| A23 | 18.3 | 13.1 | 15.4 | 17.8 | 16.7 | 18.1 | 15.9 | 18.6 | 14.9 | 13.5 | 17.5 | 15.0 | 13.9 | 14.7 | 13.6 | 17.7 | 24.6 | - | - |
| A24 | 19.0 | 12.5 | 15.8 | 19.1 | 15.6 | 18.4 | 18.0 | 17.1 | 15.3 | 12.9 | 18.4 | 15.5 | 12.6 | 17.2 | 14.7 | 15.4 | 22.3 | 12.2 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olech, M.; Kuźmak, J. Molecular Characterization of Small Ruminant Lentiviruses in Polish Mixed Flocks Supports Evidence of Cross Species Transmission, Dual Infection, a Recombination Event, and Reveals the Existence of New Subtypes within Group A. Viruses 2021, 13, 2529. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122529
Olech M, Kuźmak J. Molecular Characterization of Small Ruminant Lentiviruses in Polish Mixed Flocks Supports Evidence of Cross Species Transmission, Dual Infection, a Recombination Event, and Reveals the Existence of New Subtypes within Group A. Viruses. 2021; 13(12):2529. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122529
Chicago/Turabian StyleOlech, Monika, and Jacek Kuźmak. 2021. "Molecular Characterization of Small Ruminant Lentiviruses in Polish Mixed Flocks Supports Evidence of Cross Species Transmission, Dual Infection, a Recombination Event, and Reveals the Existence of New Subtypes within Group A" Viruses 13, no. 12: 2529. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122529