
LABRADOR—A Computational Workflow for Virus Detection in High-Throughput Sequencing Data

Abstract
:1. Introduction
2. Materials and Methods
2.1. Software Environment
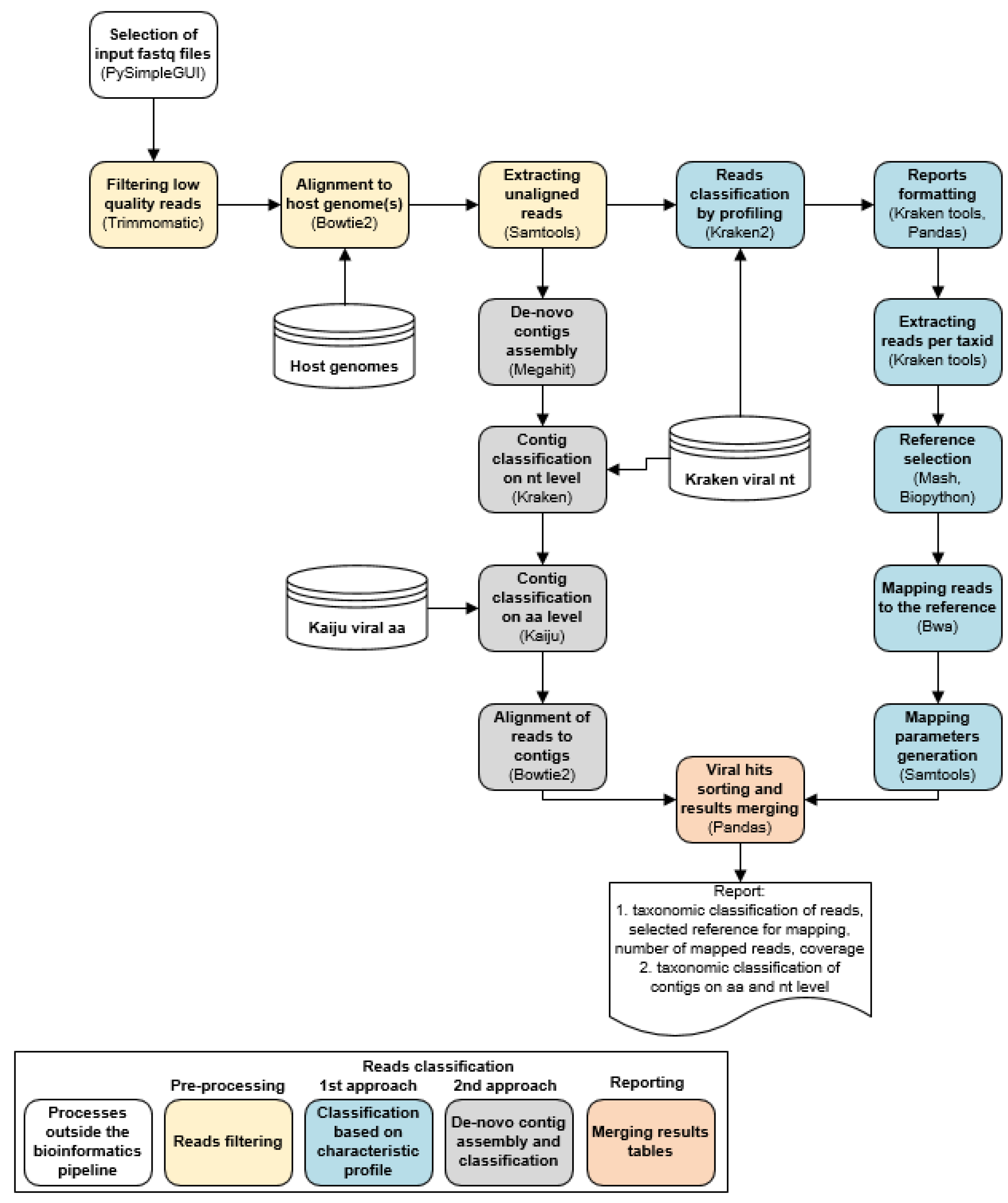
2.2. LABRADOR Wrapper
2.3. Preprocessing
2.4. Classfication of Viral Sequencing Reads and Mapping to Reference Genome
2.5. De Novo Contig Assembly and Classification
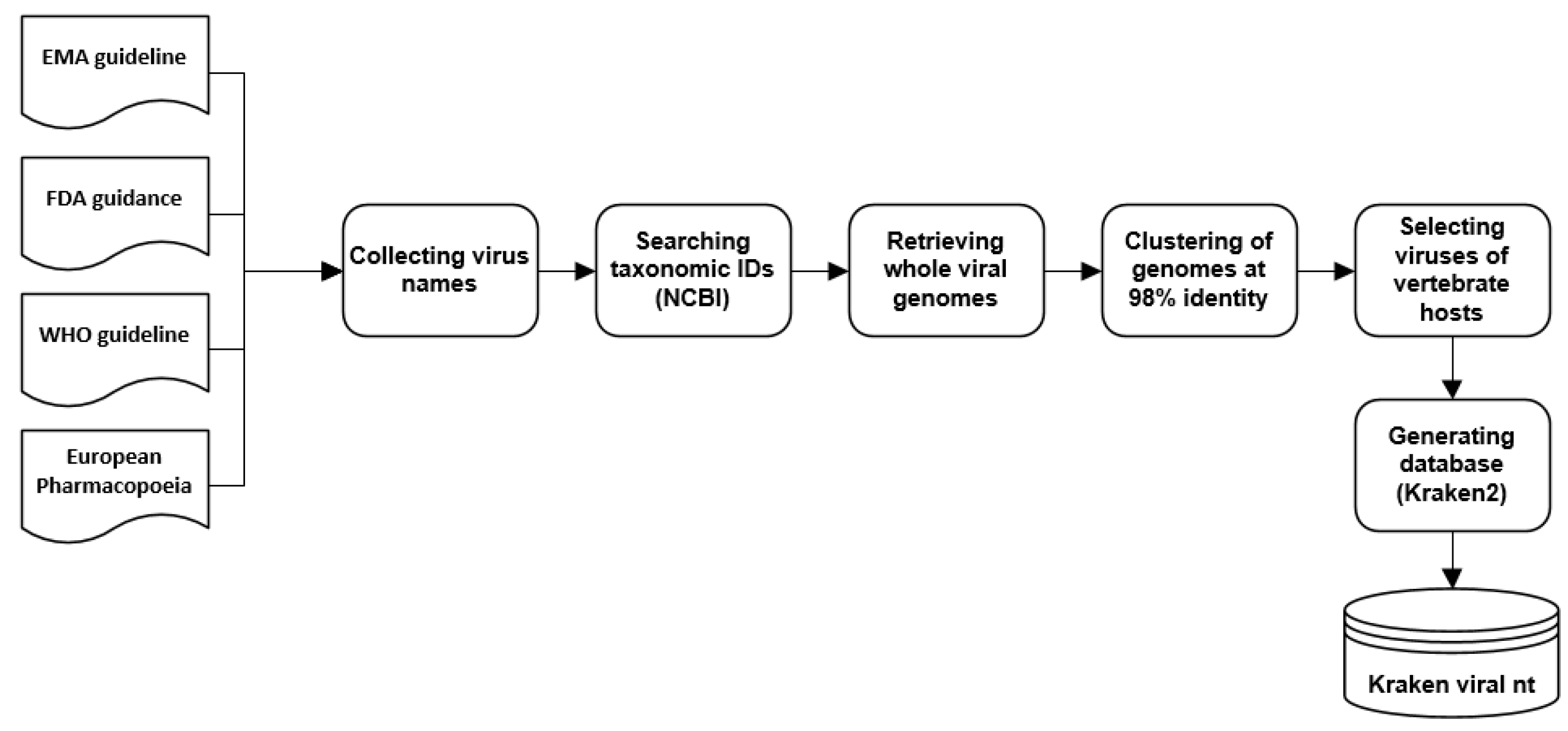
2.6. Creation of a Custom Viral Database
2.7. Evaluation of LABRADOR Workflow on Published Dataset
3. Results
3.1. Analyses Perfomed in LABRADOR Workflow and Custom Database Constuction
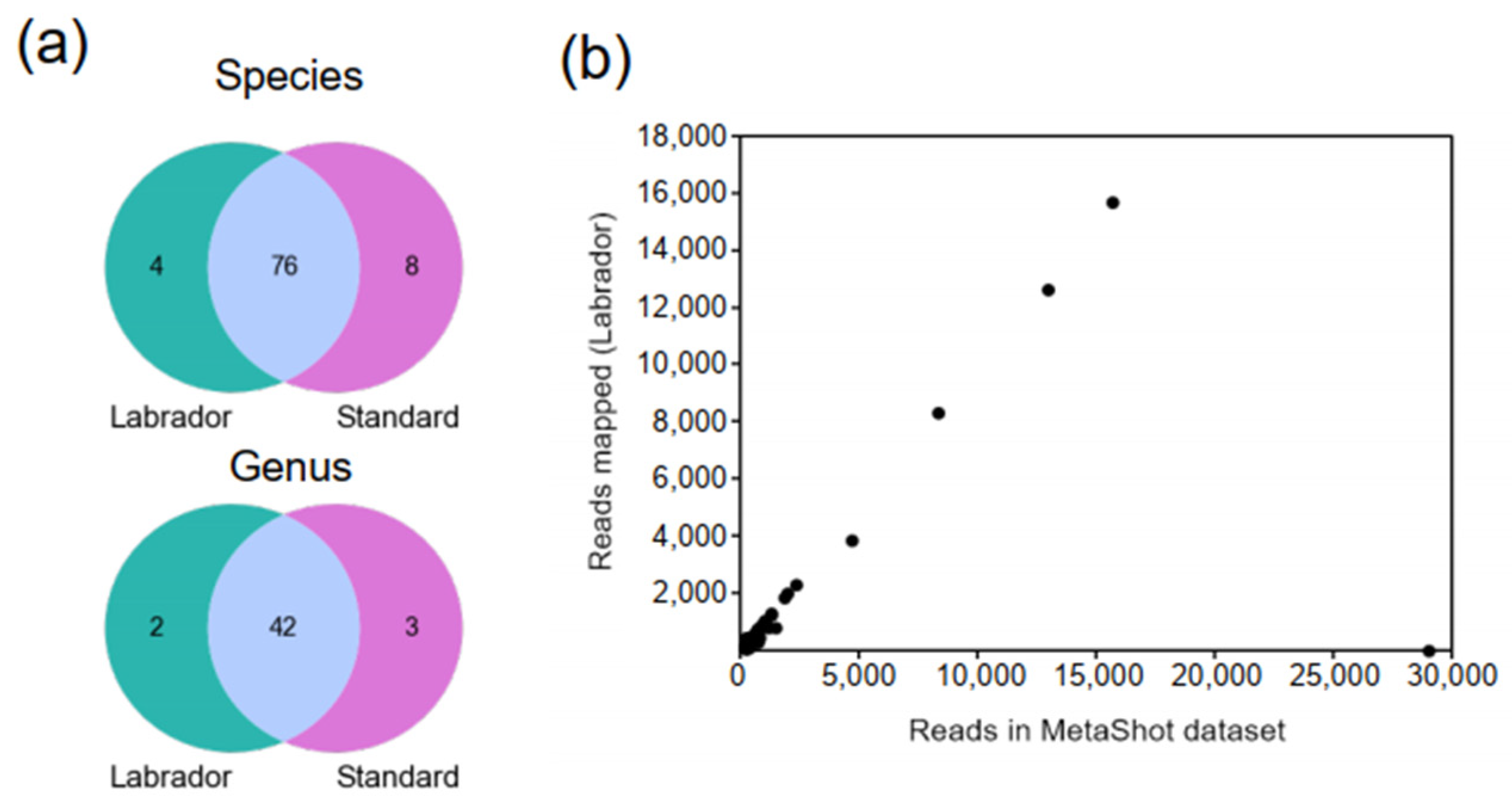
3.2. LABRADOR’s Performance on Simulated Metagenomics Dataset
3.3. Detection of AVT Model Viruses from Spiking Study
3.4. Detection of Viruses in Datasets from Real-Life Experiments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gilliland, S.M.; Forrest, L.; Carre, H.; Jenkins, A.; Berry, N.; Martin, J.; Minor, P.; Schepelmann, S. Investigation of porcine circovirus contamination in human vaccines. Biologicals 2012, 40, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Mallet, L.; Gisonni-Lex, L. Need for new technologies for detection of adventitious agents in vaccines and other biological products. PDA J. Pharm. Sci. Technol. 2014, 68, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Merten, O.W. Virus contaminations of cell cultures—A biotechnological view. Cytotechnology 2002, 39, 91–116. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Chen, E.C.; Sittler, T.; Scheinerman, A.; Roubinian, N.; Yu, G.; Kim, E.; Pillai, D.R.; Guyard, C.; Mazzulli, T.; et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE 2010, 5, e13381. [Google Scholar] [CrossRef]
- Modrof, J.; Berting, A.; Kreil, T.R. Parallel evaluation of broad virus detection methods. PDA J. Pharm. Sci. Technol. 2014, 68, 572–578. [Google Scholar] [CrossRef]
- Mee, E.T.; Preston, M.D.; Minor, P.D.; Schepelmann, S. Development of a candidate reference material for adventitious virus detection in vaccine and biologicals manufacturing by deep sequencing. Vaccine 2016, 34, 2035–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victoria, J.G.; Wang, C.; Jones, M.S.; Jaing, C.; McLoughlin, K.; Gardner, S.; Delwart, E.L. Viral nucleic acids in live-attenuated vaccines: Detection of minority variants and an adventitious virus. J. Virol. 2010, 84, 6033–6040. [Google Scholar] [CrossRef] [Green Version]
- Petricciani, J.; Sheets, R.; Griffiths, E.; Knezevic, I. Adventitious agents in viral vaccines: Lessons learned from 4 case studies. Biologicals 2014, 42, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, P.W.; Wiebe, M.E.; Leung, J.C.; Hussein, I.T.M.; Keumurian, F.J.; Bouressa, J.; Brussel, A.; Chen, D.; Chong, M.; Dehghani, H.; et al. Viral contamination in biologic manufacture and implications for emerging therapies. Nat. Biotechnol. 2020, 38, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Blümel, J.; Deforce, D.; Gruber, M.F.; Jungbäck, C.; Knezevic, I.; Mallet, L.; Mackay, D.; Matthijnssens, J.; O’Leary, M.; et al. Report of the second international conference on next generation sequencing for adventitious virus detection in biologics for humans and animals. Biologicals 2020, 67, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Frommolt, P. Challenges in the Setup of Large-scale Next-Generation Sequencing Analysis Workflows. Comput. Struct. Biotechnol. J. 2017, 15, 471–477. [Google Scholar] [CrossRef]
- Chen, S.; Huang, T.; Zhou, Y.; Han, Y.; Xu, M.; Gu, J. AfterQC: Automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinform. 2017, 18, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yan, Z.; Wu, C.; Yang, Y.; Li, X.; Zhang, G. FastProNGS: Fast preprocessing of next-generation sequencing reads. BMC Bioinform. 2019, 20, 345. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-H.; Hao, S.; Alarcon, S.; Yang, J. Comparisons of classification methods for viral genomes and protein families using alignment-free vectorization. Stat. Appl. Genet. Mol. Biol. 2018, 17. [Google Scholar] [CrossRef]
- Borozan, I.; Watt, S.N.; Ferretti, V. Evaluation of alignment algorithms for discovery and identification of pathogens using RNA-Seq. PLoS ONE 2013, 8, e76935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musich, R.; Cadle-Davidson, L.; Osier, M.V. Comparison of Short-Read Sequence Aligners Indicates Strengths and Weaknesses for Biologists to Consider. Front. Plant Sci. 2021, 12, 657240. [Google Scholar] [CrossRef]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ames, S.K.; Hysom, D.A.; Gardner, S.N.; Lloyd, G.S.; Gokhale, M.B.; Allen, J.E. Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics 2013, 29, 2253–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking metagenomics tools for taxonomic classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Gleizes, A.; Laubscher, F.; Guex, N.; Iseli, C.; Junier, T.; Cordey, S.; Fellay, J.; Xenarios, I.; Kaiser, L.; Le Mercier, P. Virosaurus A Reference to Explore and Capture Virus Genetic Diversity. Viruses 2020, 12, 1248. [Google Scholar] [CrossRef]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of virus metagenomic classification methods and their biological applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Bigot, T.; Temmam, S.; Pérot, P.; Eloit, M. RVDB-prot, a reference viral protein database and its HMM profiles. F1000Res 2019, 8, 530. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef] [Green Version]
- Fosso, B.; Santamaria, M.; D’Antonio, M.; Lovero, D.; Corrado, G.; Vizza, E.; Passaro, N.; Garbuglia, A.R.; Capobianchi, M.R.; Crescenzi, M.; et al. MetaShot: An accurate workflow for taxon classification of host-associated microbiome from shotgun metagenomic data. Bioinformatics 2017, 33, 1730–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampelli, S.; Soverini, M.; Turroni, S.; Quercia, S.; Biagi, E.; Brigidi, P.; Candela, M. ViromeScan: A new tool for metagenomic viral community profiling. BMC Genom. 2016, 17, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Ojesina, A.I.; Pedamallu, C.S.; Jung, J.; Verhaak, R.G.W.; Getz, G.; Meyerson, M. PathSeq: Software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 2011, 29, 393–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Krishnamurthy, S.; Cai, Z.; Popov, V.L.; Travassos da Rosa, A.P.; Guzman, H.; Cao, S.; Virgin, H.W.; Tesh, R.B.; Wang, D. Identification of novel viruses using VirusHunter—An automated data analysis pipeline. PLoS ONE 2013, 8, e78470. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Plyusnin, I.; Kant, R.; Jääskeläinen, A.J.; Sironen, T.; Holm, L.; Vapalahti, O.; Smura, T. Novel NGS pipeline for virus discovery from a wide spectrum of hosts and sample types. Virus Evol. 2020, 6, veaa091. [Google Scholar] [CrossRef]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Braxton, C.; Charlebois, R.L.; Deyati, A.; Duncan, P.; La Neve, F.; Malicki, H.D.; Ribrioux, S.; Rozelle, D.K.; Michaels, B.; et al. Considerations for optimization of high-throughput sequencing bioinformatics pipelines for virus detection. Viruses 2018, 10, 528. [Google Scholar] [CrossRef] [Green Version]
- Sutton, T.D.S.; Clooney, A.G.; Ryan, F.J.; Ross, R.P.; Hill, C. Choice of assembly software has a critical impact on virome characterisation. Microbiome 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking viromics: An in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5, e3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Walt, A.J.; van Goethem, M.W.; Ramond, J.-B.; Makhalanyane, T.P.; Reva, O.; Cowan, D.A. Assembling metagenomes, one community at a time. BMC Genom. 2017, 18, 521. [Google Scholar] [CrossRef] [PubMed]
- Orton, R.J.; Gu, Q.; Hughes, J.; Maabar, M.; Modha, S.; Vattipally, S.B.; Wilkie, G.S.; Davison, A.J. Bioinformatics tools for analysing viral genomic data. Rev. Sci. Tech. 2016, 35, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Angle, C.; Waggoner, L.P.; Ferrando, A.; Haney, P.; Passler, T. Canine Detection of the Volatilome: A Review of Implications for Pathogen and Disease Detection. Front. Vet. Sci. 2016, 3, 47. [Google Scholar] [CrossRef]
- Jendrny, P.; Schulz, C.; Twele, F.; Meller, S.; von Köckritz-Blickwede, M.; Osterhaus, A.D.M.E.; Ebbers, J.; Pilchová, V.; Pink, I.; Welte, T.; et al. Scent dog identification of samples from COVID-19 patients—A pilot study. BMC Infect. Dis. 2020, 20, 536. [Google Scholar] [CrossRef]
- EMA. Guideline on Requirements for the Production and Control of Immunological Veterinary Medicinal Products. 2016. Available online: https://www.ema.europa.eu/en/requirements-production-control-immunological-veterinary-medicinal-products (accessed on 2 March 2020).
- European Directorate for the Quality of Medicines & HealthCare. Cell Substrates for the Production of Vaccines for Human Use. In European Pharmacopoeia, 9th ed; Council of Europe: Strasbourg, France, 2017. [Google Scholar]
- FDA. Guidance for Industry—Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications. 2010. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/characterization-and-qualification-cell-substrates-and-other-biological-materials-used-production (accessed on 2 March 2020).
- WHO. Recommendations for the Evaluation of Animal Cell Cultures as Substrates for the Manufacture of Biological Medicinal Products and for the Characterization of Cell Banks; WHO Technical Report Series, No. 978; WHO Press World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondov, B.D.; Starrett, G.J.; Sappington, A.; Kostic, A.; Koren, S.; Buck, C.B.; Phillippy, A.M. Mash Screen: High-throughput sequence containment estimation for genome discovery. Genome Biol. 2019, 20, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Dwivedi, B.; Akhter, S.; Breitbart, M.; Edwards, R.A. Multidimensional metrics for estimating phage abundance, distribution, gene density, and sequence coverage in metagenomes. Front. Microbiol. 2015, 6, 381. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-M.; Luo, R.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Hulo, C.; de Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.S.; Ng, S.H.S.; Vandeputte, O.; Aljanahi, A.; Deyati, A.; Cassart, J.-P.; Charlebois, R.L.; Taliaferro, L.P. A multicenter study to evaluate the performance of high-throughput sequencing for virus detection. mSphere 2018, 2, e00307-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar] [CrossRef]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Yi, X.; Zhuang, Y. Coupling metabolomics analysis and DOE optimization strategy towards enhanced IBDV production by chicken embryo fibroblast DF-1 cells. J. Biotechnol. 2020, 307, 114–124. [Google Scholar] [CrossRef]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roser, L.G.; Agüero, F.; Sánchez, D.O. FastqCleaner: An interactive Bioconductor application for quality-control, filtering and trimming of FASTQ files. BMC Bioinform. 2019, 20, 361. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Muller, J.; Sears, J.F. Early detection of endogenous retroviruses in chemically induced mouse cells. Virus Res. 2001, 79, 39–45. [Google Scholar] [CrossRef]
- Vollmers, J.; Wiegand, S.; Kaster, A.-K. Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective—Not only size matters! PLoS ONE 2017, 12, e0169662. [Google Scholar] [CrossRef] [Green Version]
- Lapidus, A.L.; Korobeynikov, A.I. Metagenomic data assembly—The way of decoding unknown microorganisms. Front. Microbiol. 2021, 12, 613791. [Google Scholar] [CrossRef] [PubMed]
- Charlebois, R.L.; Sathiamoorthy, S.; Logvinoff, C.; Gisonni-Lex, L.; Mallet, L.; Ng, S.H.S. Sensitivity and breadth of detection of high-throughput sequencing for adventitious virus detection. NPJ Vaccines 2020, 5, 1926. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Taxomic Level | Tool | Precision [%] | Recall [%] |
|---|---|---|---|
| Species | LABRADOR | 95 | 90.5 |
| Lazypipe-nt | 97.2 | 82.1 | |
| Lazypipe | 90.0 | 85.7 | |
| Centifuge | 63.0 | 95.2 | |
| MetaPhlan2 | 84.4 | 45.2 | |
| Kraken2 | 94.1 | 19.0 | |
| Genus | LABRADOR | 95.5 | 93.3 |
| Lazypipe-nt | 95.3 | 91.1 | |
| Lazypipe | 95.3 | 91.1 | |
| Centifuge | 84.9 | 100 | |
| MetaPhlan2 | 88.9 | 71.1 | |
| Kraken2 | 95.5 | 46.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabiańska, I.; Borutzki, S.; Richter, B.; Tran, H.Q.; Neubert, A.; Mayer, D. LABRADOR—A Computational Workflow for Virus Detection in High-Throughput Sequencing Data. Viruses 2021, 13, 2541. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122541
Fabiańska I, Borutzki S, Richter B, Tran HQ, Neubert A, Mayer D. LABRADOR—A Computational Workflow for Virus Detection in High-Throughput Sequencing Data. Viruses. 2021; 13(12):2541. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122541
Chicago/Turabian StyleFabiańska, Izabela, Stefan Borutzki, Benjamin Richter, Hon Q. Tran, Andreas Neubert, and Dietmar Mayer. 2021. "LABRADOR—A Computational Workflow for Virus Detection in High-Throughput Sequencing Data" Viruses 13, no. 12: 2541. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122541