
Exploring the Human Cytomegalovirus Core Nuclear Egress Complex as a Novel Antiviral Target: A New Type of Small Molecule Inhibitors



Abstract
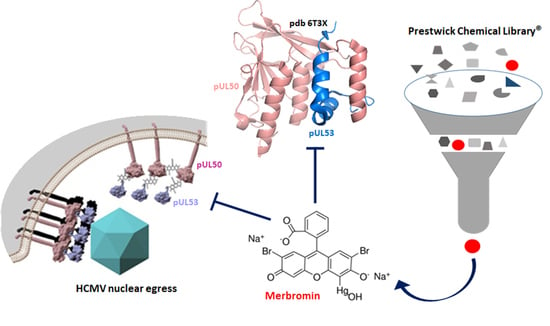
:
1. Introduction
2. Materials and Methods
2.1. pUL50-pUL53 Inhibition Assay
2.2. HIV-1 gp120–mAb 447-52D Inhibition Assay
2.3. Cytotoxicity Assay
2.4. Virus Infection, Plaque Reduction, and Viral Yield Assays
2.5. Quantitative Polymerase Chain Reaction (qPCR)
2.6. Indirect Immunofluorescence Assay and Confocal Laser-Scanning Microscopy
2.7. Antibodies
3. Results and Discussion
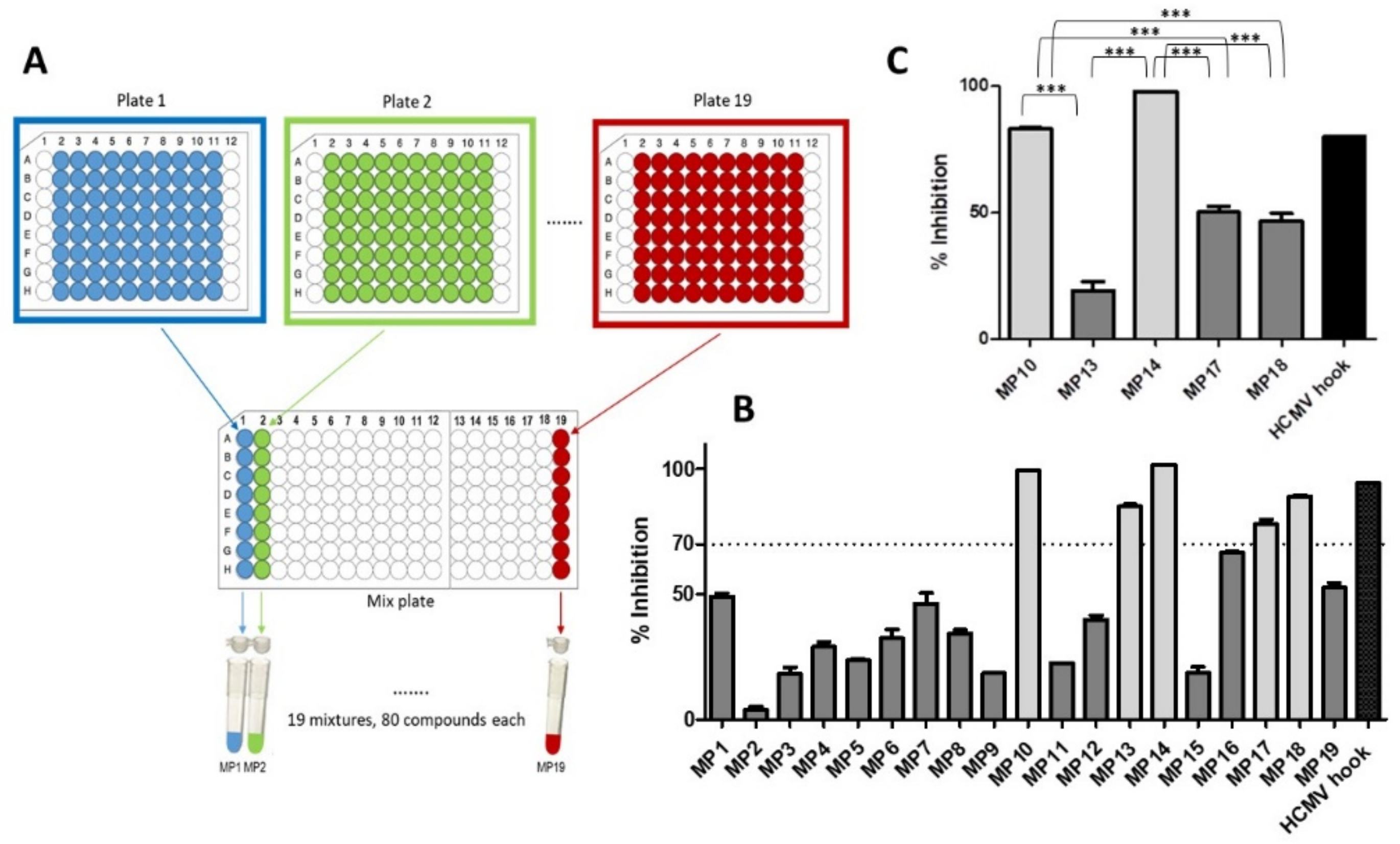
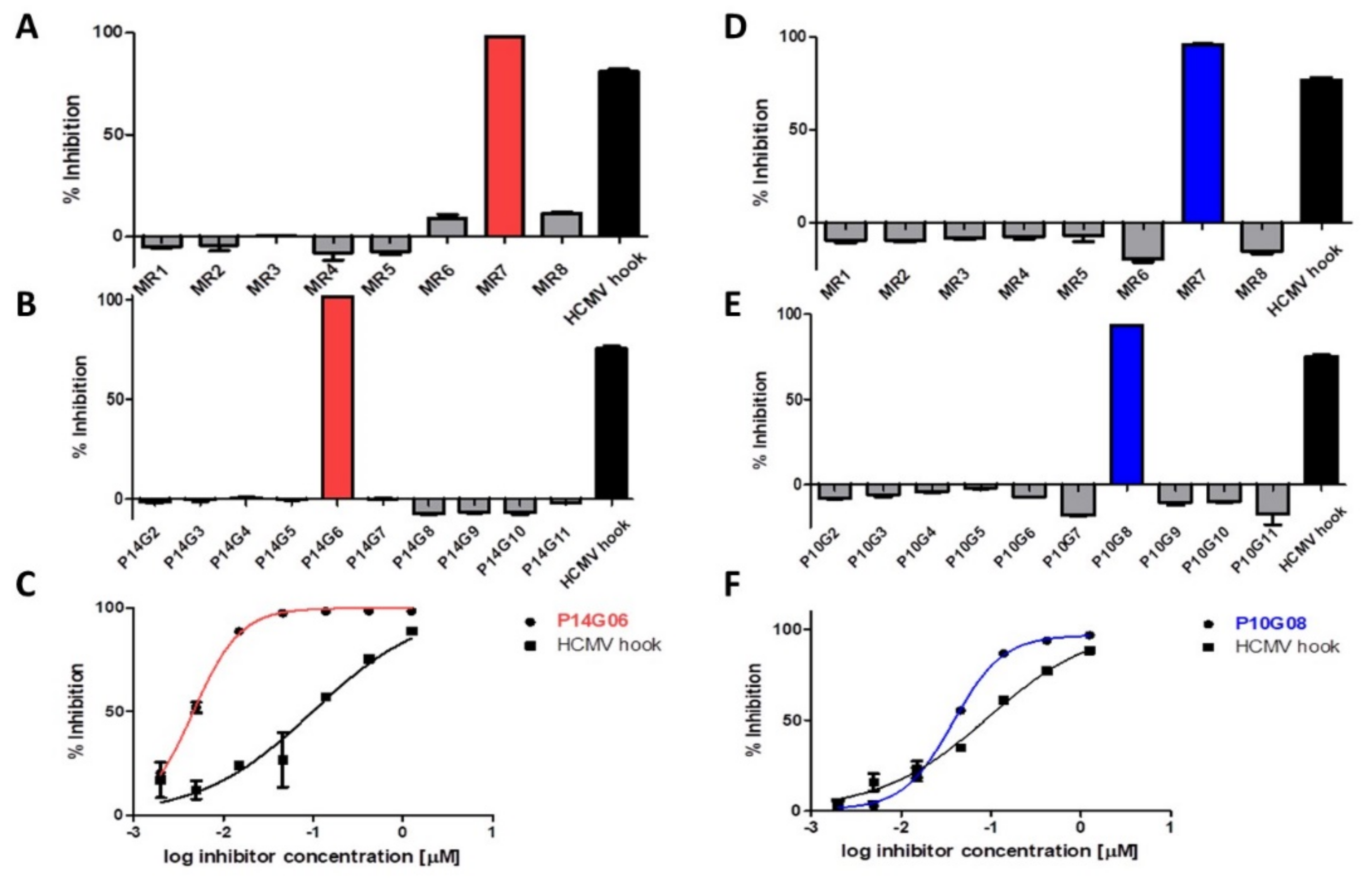
3.1. Identification of HCMV Core NEC Inhibitors from the Prestwick Chemical Library®
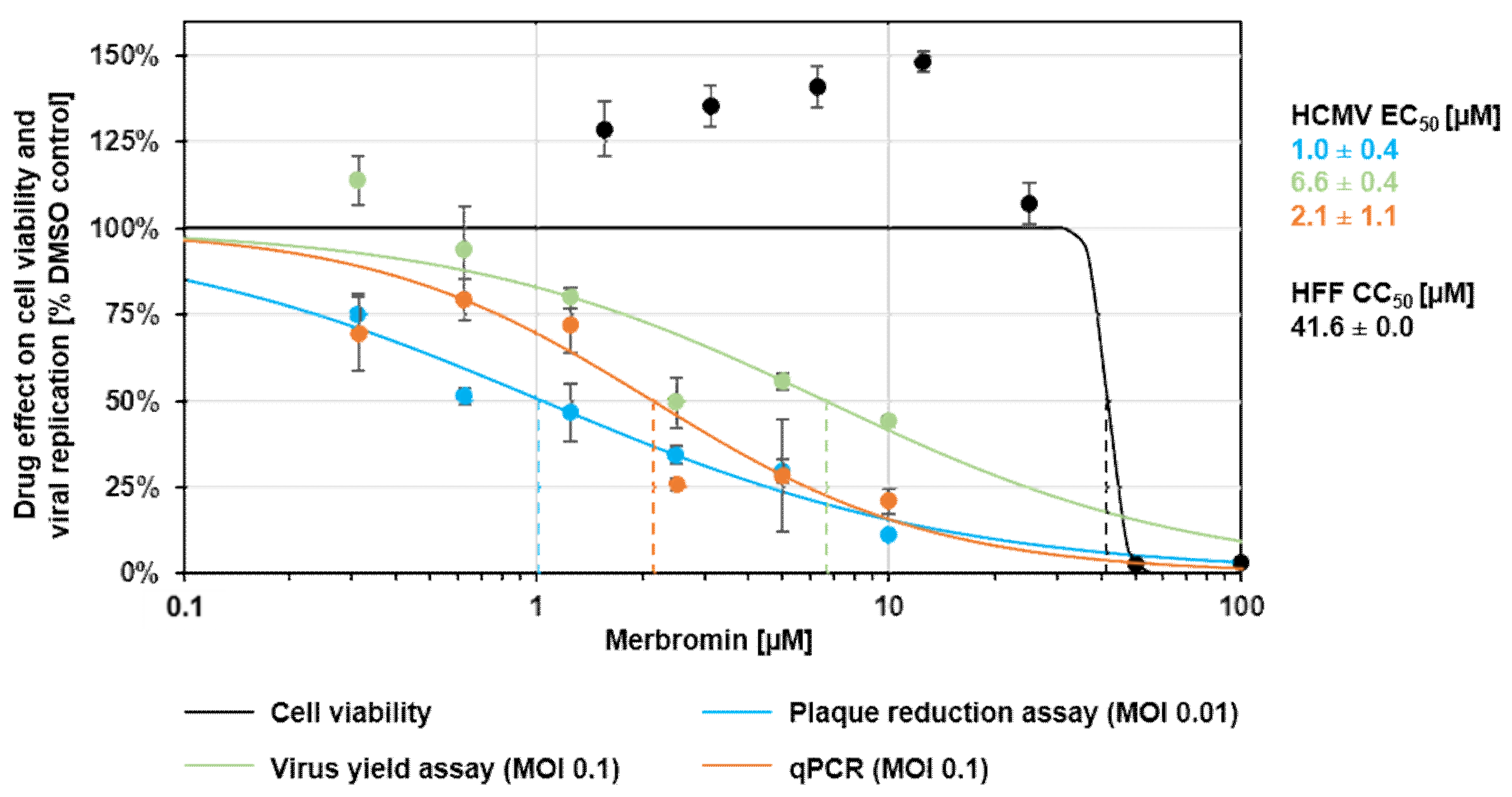
3.2. Cytotoxicity and Selectivity of Merbromin
3.3. Antiviral Activity of Merbromin
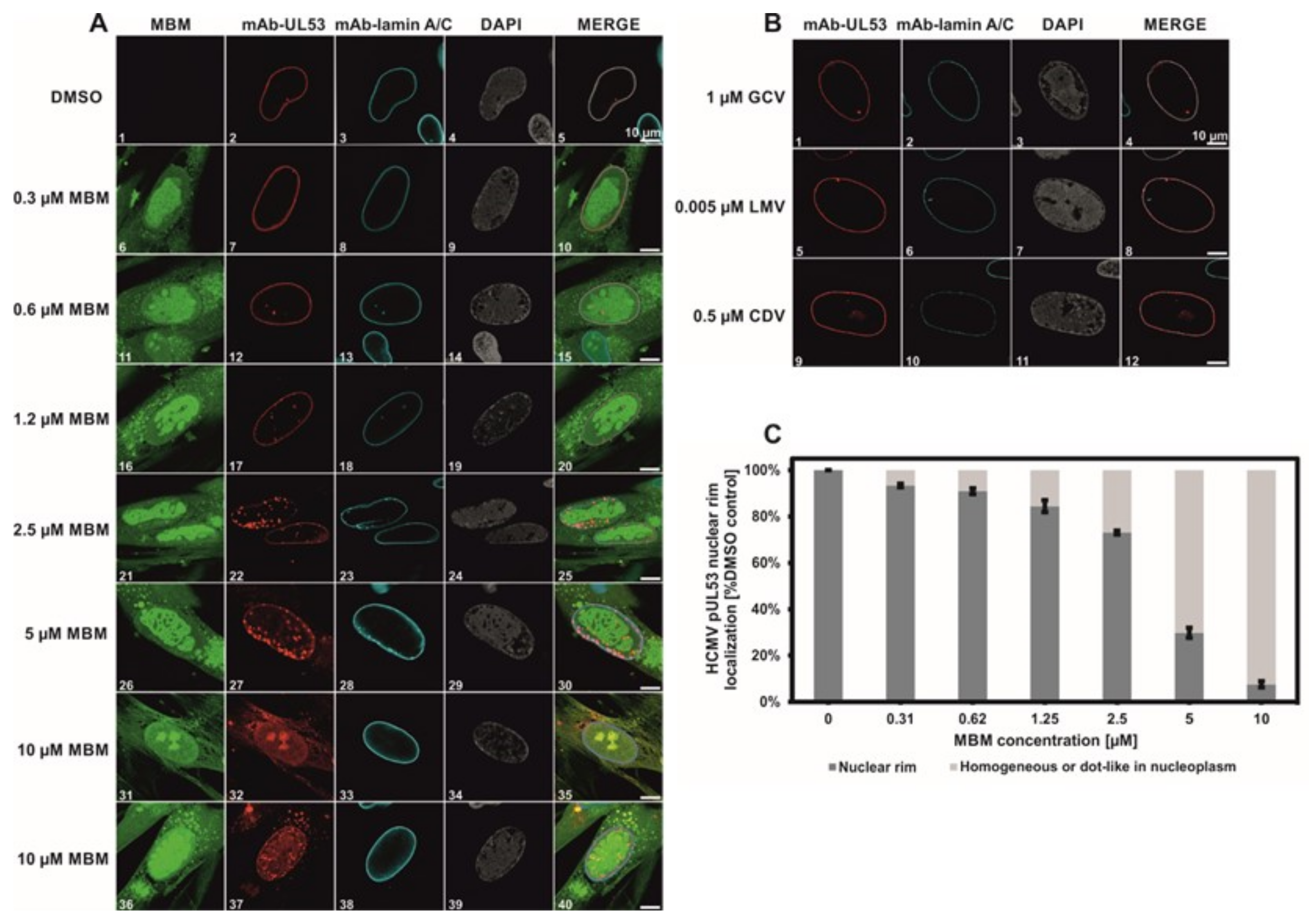
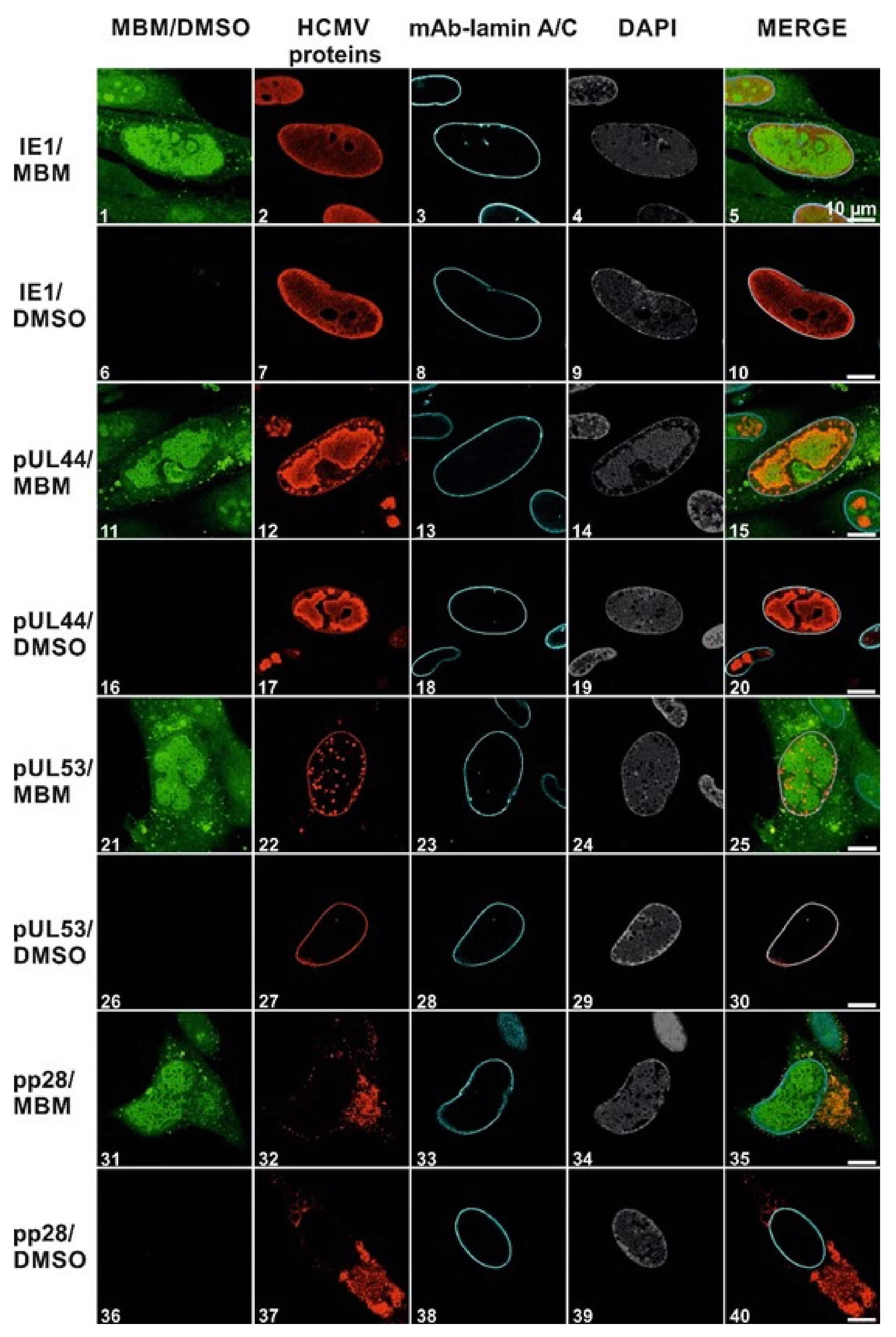
3.4. Selective Effect of Merbromin on the HCMV Core NEC
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andrei, G.; De Clercq, E.; Snoeck, R. Drug targets in cytomegalovirus infection. Infect. Disord. Drug Targets 2009, 9, 201–222. [Google Scholar] [CrossRef]
- Kawasaki, H.; Kosugi, I.; Meguro, S.; Iwashita, T. Pathogenesis of developmental anomalies of the central nervous system induced by congenital cytomegalovirus infection. Pathol. Int. 2017, 67, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J.; Prichard, M.N. New therapies for human cytomegalovirus infections. Antivir. Res. 2018, 159, 153–174. [Google Scholar] [CrossRef]
- Steingruber, M.; Marschall, M. The Cytomegalovirus Protein Kinase pUL97: Host Interactions, Regulatory Mechanisms and Antiviral Drug Targeting. Microorganisms 2020, 8, 515. [Google Scholar] [CrossRef] [Green Version]
- Mercorelli, B.; Luganini, A.; Celegato, M.; Palù, G.; Gribaudo, G.; Lepesheva, G.I.; Loregian, A. The Clinically Approved Antifungal Drug Posaconazole Inhibits Human Cytomegalovirus Replication. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Kapoor, A.; Ghosh, A.K.; Forman, M.; Hu, X.; Ye, W.; Southall, N.; Marugan, J.; Keyes, R.F.; Smith, B.C.; Meyers, D.J.; et al. Validation and Characterization of Five Distinct Novel Inhibitors of Human Cytomegalovirus. J. Med. Chem. 2020, 63, 3896–3907. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 2017, 27. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, E.; Hamilton, S.T.; Bahsi, H.; Wagner, S.; Jonjic, S.; Rawlinson, W.D.; Marschall, M.; Milbradt, J. Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J. Gen. Virol. 2016, 97, 1676–1685. [Google Scholar] [CrossRef] [Green Version]
- Milbradt, J.; Kraut, A.; Hutterer, C.; Sonntag, E.; Schmeiser, C.; Ferro, M.; Wagner, S.; Lenac, T.; Claus, C.; Pinkert, S.; et al. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol. Cell Prot. 2014, 13, 2132–2146. [Google Scholar] [CrossRef] [Green Version]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kicuntod, J.; Alkhashrom, S.; Häge, S.; Diewald, B.; Müller, R.; Hahn, F.; Lischka, P.; Sticht, H.; Eichler, J.; Marschall, M. Properties of oligomeric interaction of the cytomegalovirus core nuclear egress complex (NEC) and its sensitivity to an NEC inhibitory small molecule. Viruses 2021, 13, 462. [Google Scholar] [CrossRef]
- Muller, Y.A.; Hage, S.; Alkhashrom, S.; Hollriegl, T.; Weigert, S.; Dolles, S.; Hof, K.; Walzer, S.A.; Egerer-Sieber, C.; Conrad, M.; et al. High-resolution crystal structures of two prototypical beta- and gamma-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J. Biol. Chem. 2020, 295, 3189–3201. [Google Scholar] [CrossRef]
- Marschall, M.; Hage, S.; Conrad, M.; Alkhashrom, S.; Kicuntod, J.; Schweininger, J.; Kriegel, M.; Losing, J.; Tillmanns, J.; Neipel, F.; et al. Nuclear Egress Complexes of HCMV and Other Herpesviruses: Solving the Puzzle of Sequence Coevolution, Conserved Structures and Subfamily-Spanning Binding Properties. Viruses 2020, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [Green Version]
- Schnee, M.; Wagner, F.M.; Koszinowski, U.H.; Ruzsics, Z. A cell free protein fragment complementation assay for monitoring the core interaction of the human cytomegalovirus nuclear egress complex. Antiviral. Res. 2012, 95, 12–18. [Google Scholar] [CrossRef]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Liu, N.; Sheng, C. Allosteric Modulators of Protein-Protein Interactions (PPIs). Adv. Exp. Med. Biol. 2019, 1163, 313–334. [Google Scholar] [CrossRef]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Häge, S.; Kraut, A.; Hesse, A.-M.; Amin, B.; Sonnewald, U.; Couté, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef]
- Scott, L.J.; Goa, K.L. Verteporfin. Drugs Aging 2000, 16, 139–146; discussion 147–148. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Li, X. Verteporfin inhibits cell proliferation and induces apoptosis in different subtypes of breast cancer cell lines without light activation. BMC Cancer 2020, 20, 1042. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.W.; Xiao, S.; Ogomori, K.; Hammarstedt, J.E.; Little, E.C.; Lang, D. The Efficiency of Verteporfin as a Therapeutic Option in Pre-Clinical Models of Melanoma. J. Cancer 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Norman, G.; Christie, J.; Liu, Z.; Westby, M.J.; Jefferies, J.M.; Hudson, T.; Edwards, J.; Mohapatra, D.P.; Hassan, I.A.; Dumville, J.C. Antiseptics for burns. Cochrane Database Syst. Rev. 2017, 7, CD011821. [Google Scholar] [CrossRef] [PubMed]
- Vachhrajani, V.; Khakhkhar, P. Antiseptics and Local Antibiotics. In Science of Wound Healing and Dressing Materials; Vachhrajani, V., Khakhkhar, P., Eds.; Springer Singapore: Singapore, 2020; pp. 43–57. [Google Scholar]
- Ajsuvakova, O.P.; Tinkov, A.A.; Aschner, M.; Rocha, J.B.T.; Michalke, B.; Skalnaya, M.G.; Skalny, A.V.; Butnariu, M.; Dadar, M.; Sarac, I.; et al. Sulfhydryl groups as targets of mercury toxicity. Coord. Chem. Rev. 2020, 417, 213343. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pang, G.; Zhao, D.; Gao, C.; Zhou, L.; Sun, S.; Wang, B. In vitro activity of thimerosal against ocular pathogenic fungi. Antimicrob. Agents Chemother. 2010, 54, 536–539. [Google Scholar] [CrossRef] [Green Version]
- Robbins, E.B.; Chen, K.K. A new mercurial diuretic. J. Am. Pharm. Assoc. 1951, 40, 249–251. [Google Scholar] [CrossRef]
- Gorny, M.K.; Conley, A.J.; Karwowska, S.; Buchbinder, A.; Xu, J.Y.; Emini, E.A.; Koenig, S.; Zolla-Pazner, S. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J. Virol. 1992, 66, 7538–7542. [Google Scholar] [CrossRef] [Green Version]
- Möbius, K.; Dürr, R.; Haußner, C.; Dietrich, U.; Eichler, J. A Functionally Selective Synthetic Mimic of the HIV-1 Coreceptor CXCR4. Chem. Eur. J. 2012, 18, 8292–8295. [Google Scholar] [CrossRef] [PubMed]
- Groß, A.; Möbius, K.; Haußner, C.; Donhauser, N.; Schmidt, B.; Eichler, J. Mimicking Protein–Protein Interactions through Peptide–Peptide Interactions: HIV-1 gp120 and CXCR4. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häge, S.; Sonntag, E.; Borst, E.M.; Tannig, P.; Seyler, L.; Bäuerle, T.; Bailer, S.M.; Lee, C.-P.; Müller, R.; Wangen, C.; et al. Patterns of Autologous and Nonautologous Interactions between Core Nuclear Egress Complex (NEC) Proteins of α-, β- and γ-Herpesviruses. Viruses 2020, 12, 303. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions of HCMV Infection b | TB40 UL32-GFP on MRC-5 | TB40E on ARPE-19 | AD169 on HFF | |
|---|---|---|---|---|
| DMSO | 88 ± 0.17% | 94.3 ± 2.73% | 89 ± 1.39% | |
| merbromin | 1.25 µM | 61 ± 1.24% | 79.4 ± 1.29% | 71 ± 4.28% |
| 2.5 µM | 51 ± 1.84% | 49.5 ± 0.87% | 48 ± 0.05% | |
| 5 µM | 35 ± 1.28% | 27.4 ± 2.38% | 32 ± 0.41% | |
| 10 µM | 26 ± 0.51% | 26.8 ± 0.97% | 23 ± 1.01% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhashrom, S.; Kicuntod, J.; Häge, S.; Schweininger, J.; Muller, Y.A.; Lischka, P.; Marschall, M.; Eichler, J. Exploring the Human Cytomegalovirus Core Nuclear Egress Complex as a Novel Antiviral Target: A New Type of Small Molecule Inhibitors. Viruses 2021, 13, 471. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030471
Alkhashrom S, Kicuntod J, Häge S, Schweininger J, Muller YA, Lischka P, Marschall M, Eichler J. Exploring the Human Cytomegalovirus Core Nuclear Egress Complex as a Novel Antiviral Target: A New Type of Small Molecule Inhibitors. Viruses. 2021; 13(3):471. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030471
Chicago/Turabian StyleAlkhashrom, Sewar, Jintawee Kicuntod, Sigrun Häge, Johannes Schweininger, Yves A. Muller, Peter Lischka, Manfred Marschall, and Jutta Eichler. 2021. "Exploring the Human Cytomegalovirus Core Nuclear Egress Complex as a Novel Antiviral Target: A New Type of Small Molecule Inhibitors" Viruses 13, no. 3: 471. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030471