
Avian Influenza A Viruses Reassort and Diversify Differently in Mallards and Mammals

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation and Propagation
2.2. Whole Genome Sequencing and Primer Design
2.3. Cells
2.4. Animal Models
2.5. Experimental Infection of Mallards
2.6. Experimental Infection of Guinea Pigs
2.7. Determination of Viral Loads
2.8. Quantification of Reassortment
2.9. Software
2.10. Genotype Frequencies, Richness, Diversity, and Evenness
2.11. Linkage Disequilibrium
2.12. Minority Segments, Unique Genotypes, and Simulation
2.13. Statistical Measures
3. Results
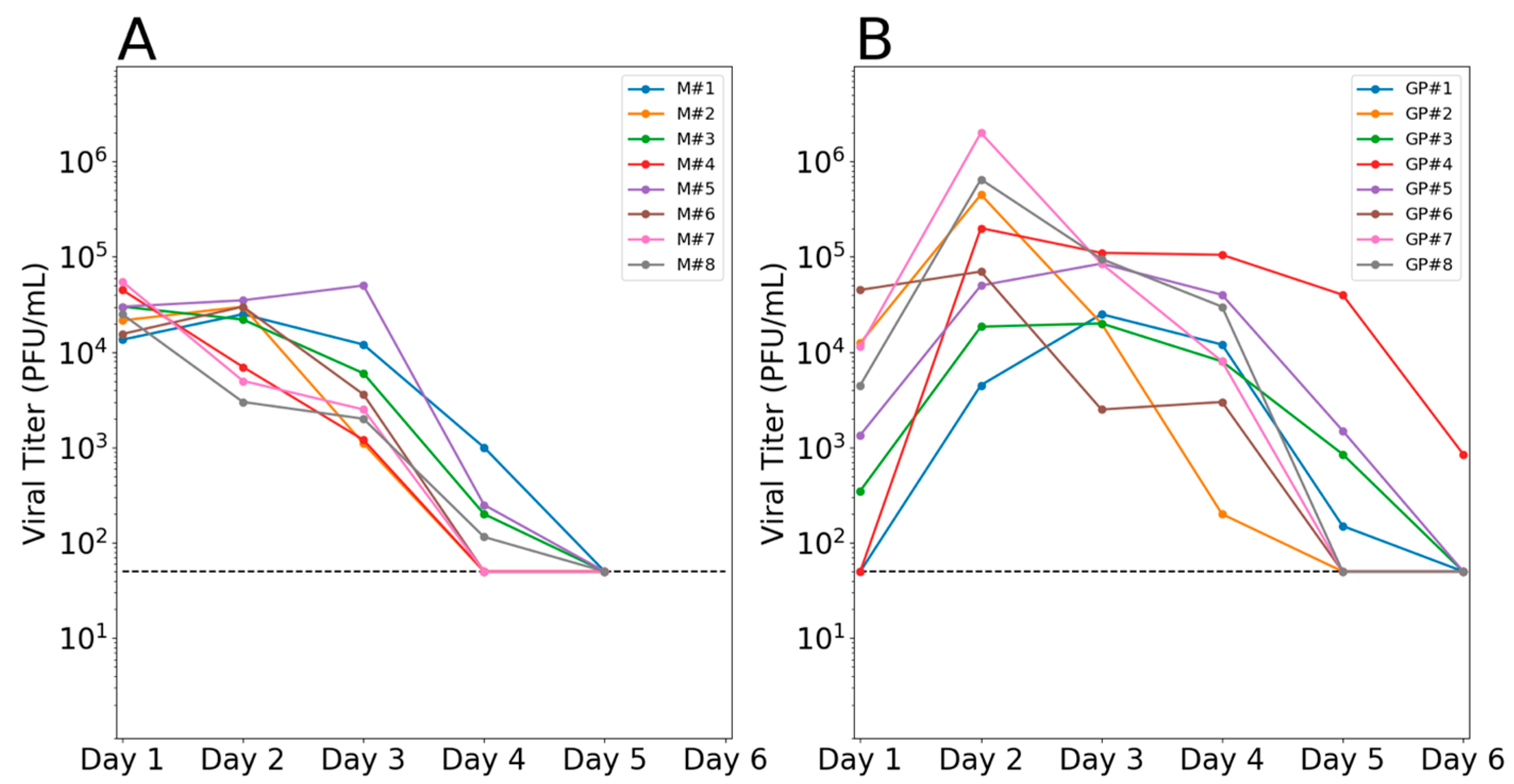
3.1. Robust Infection in Co-Inoculated Mallards and Guinea Pigs
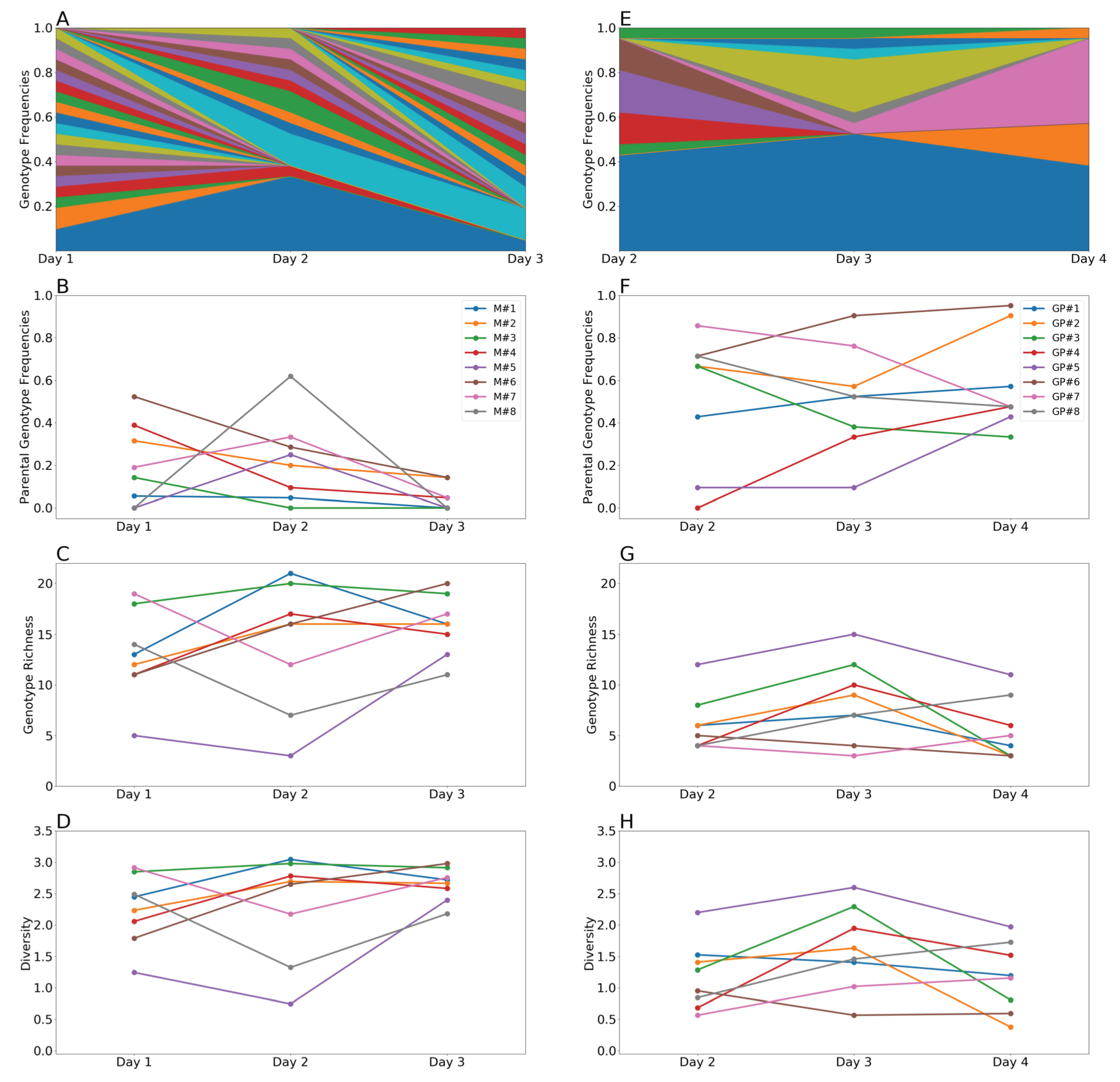
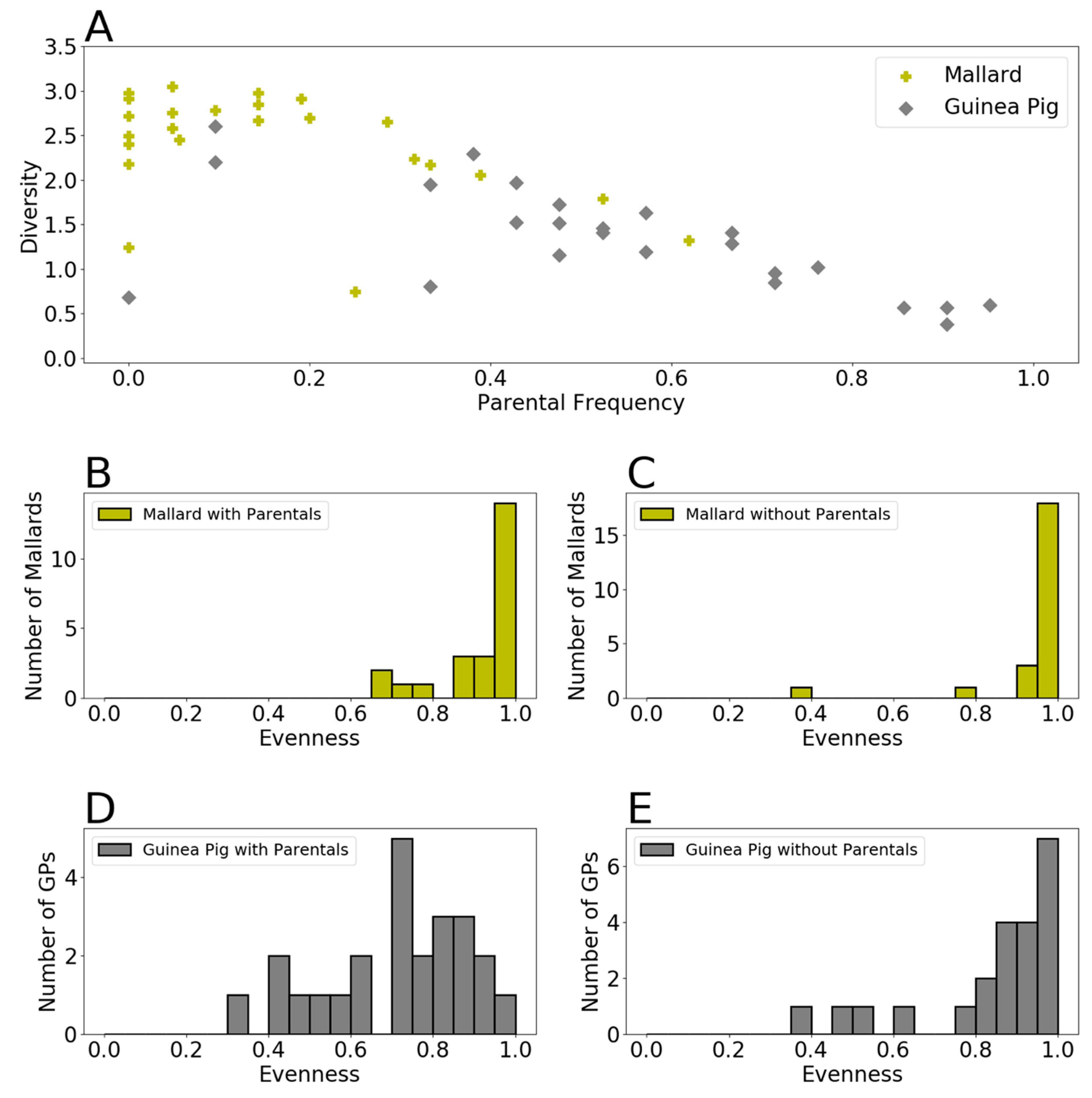
3.2. Genotypic Diversity Was Higher in Mallards Than in Guinea Pigs
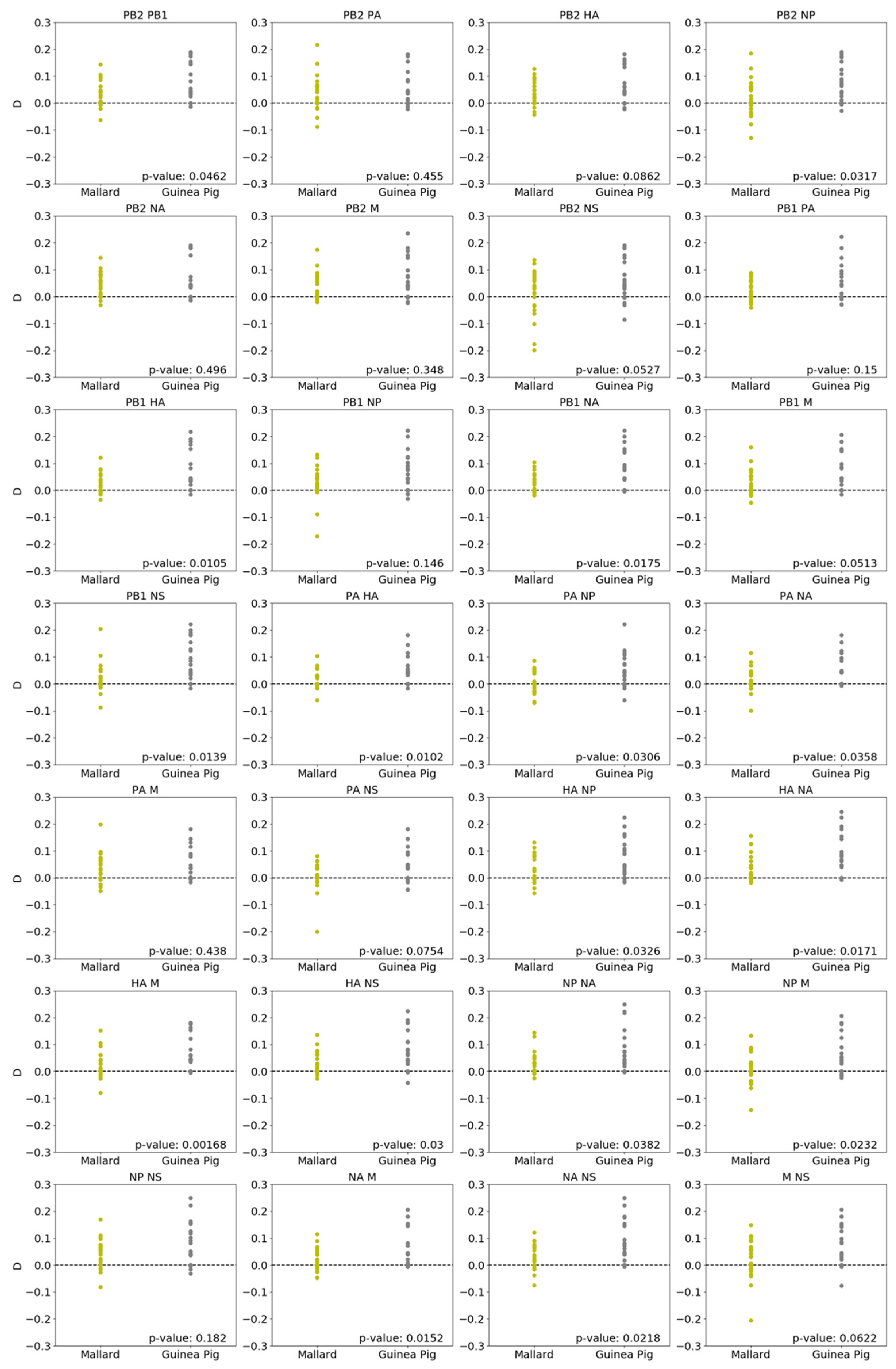
3.3. Parental Genotypes Drive Higher Linkage Disequilibrium in Guinea Pigs Than in Mallards
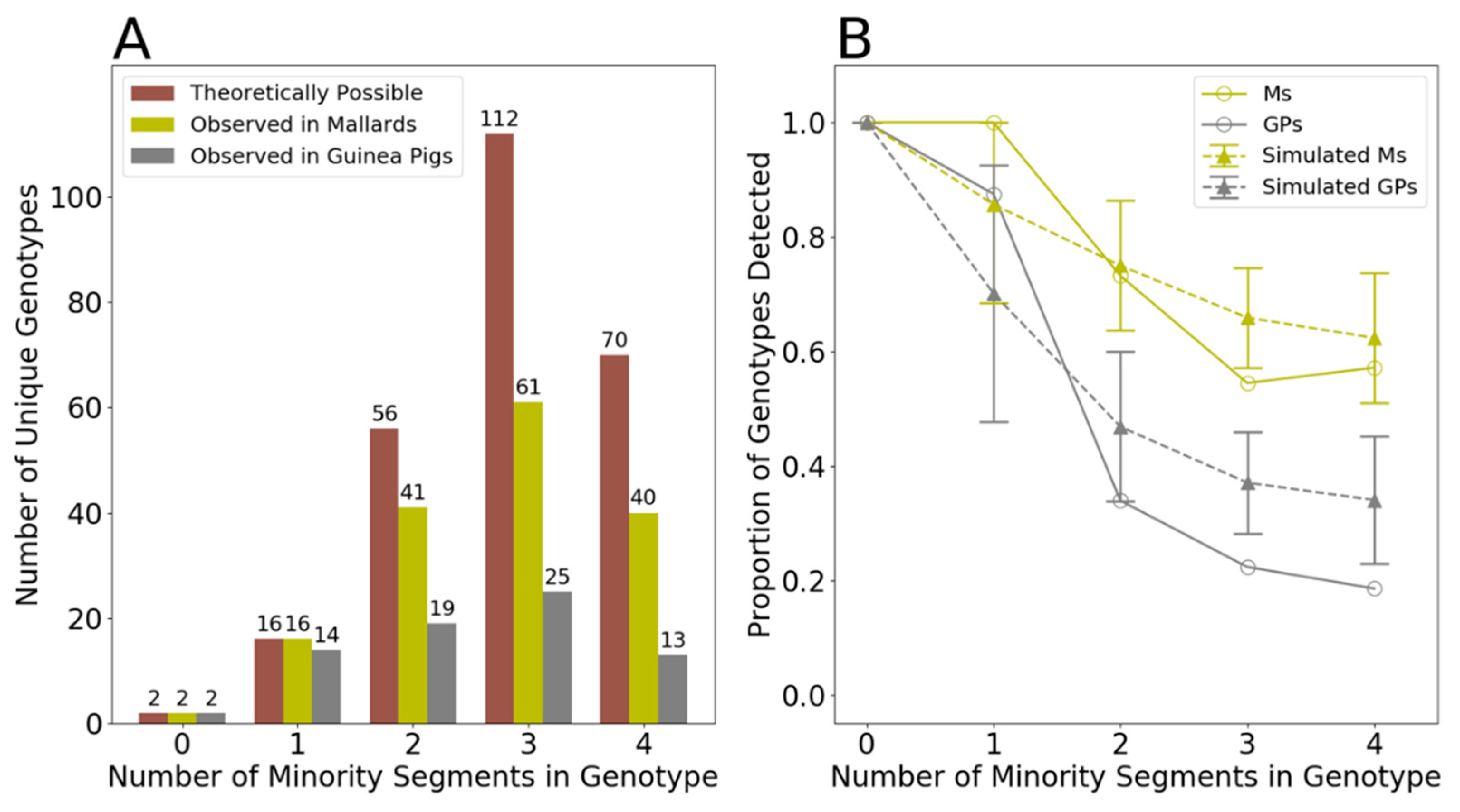
3.4. Genotype Patterns Are Consistent with Random Sampling of Gene Segments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethics Statement
References
- Webster, R.G.; Hinshaw, V.S.; Bean, W.J.; Turner, B.; Shortridge, K.F. Influenza viruses from avian and porcine sources and their possible role in the origin of human pandemic strains. Dev. Boil. Stand. 1977, 39, 461–468. [Google Scholar]
- Wright, P.F.; Neumann, G.; Kawaoka, Y. Orthomyxoviruses. In Fields Virology; Fields, B.N., Knipe, D.M., Chanock, R.M., Hirsch, M.S., Melnick, J.L., Monath, T.P., Roizman, B., Eds.; Lippincott-Raven: Pennsylvania, PA, USA, 1996; Volume 1, pp. 1691–1740. [Google Scholar]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Shortridge, K.F.; Kawaoka, Y. Influenza: Interspecies transmission and emergence of new pandemics. FEMS Immunol. Med. Microbiol. 1997, 18, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The mother of all pandemics. Emerg. Infect. Dis. 2006, 12, 69–79. [Google Scholar] [CrossRef]
- Viboud, C.; Miller, M.; Olson, D.R.; Osterholm, M.; Simonsen, L. Preliminary Estimates of Mortality and Years of Life Lost Associated with the 2009 A/H1N1 Pandemic in the US and Comparison with Past Influenza Seasons. PLoS Curr. 2010, 2, RRN1153. [Google Scholar] [CrossRef]
- Marshall, N.; Priyamvada, L.; Ende, Z.; Steel, J.; Lowen, A.C. Influenza Virus Reassortment Occurs with High Frequency in the Absence of Segment Mismatch. PLOS Pathog. 2013, 9, e1003421. [Google Scholar] [CrossRef] [Green Version]
- Phipps, K.L.; Marshall, N.; Tao, H.; Danzy, S.; Onuoha, N.; Steel, J.; Lowen, A.C. Seasonal H3N2 and 2009 Pandemic H1N1 Influenza A Viruses Reassort Efficiently but Produce Attenuated Progeny. J. Virol. 2017, 91, e00830-17. [Google Scholar] [CrossRef] [Green Version]
- Villa, M.; Lässig, M. Fitness cost of reassortment in human influenza. PLoS Pathog. 2017, 13, e1006685. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.; Van Hoeven, N.; Chen, L.-M.; Maines, T.R.; Cox, N.J.; Katz, J.M.; Donis, R.O. Reassortment between Avian H5N1 and Human H3N2 Influenza Viruses in Ferrets: A Public Health Risk Assessment. J. Virol. 2009, 83, 8131–8140. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.J.; Hill, N.J.; Zabilansky, J.; Yuan, K.; Runstadler, J.A. Reticulate evolution is favored in influenza niche switching. Proc. Natl. Acad. Sci. USA 2016, 113, 5335–5339. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.T.-Y.; Wang, J.; Shen, Y.; Zhou, B.; Duan, L.; Cheung, C.-L.; Ma, C.; Lycett, S.J.; Leung, C.Y.-H.; Chen, X.; et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nat. Cell Biol. 2013, 502, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Su, C.; Wang, D.; Peng, Y.; Liu, M.; Hua, S.; Li, T.; Gao, G.F.; Tang, H.; Chen, J.; et al. Sequential Reassortments Underlie Diverse Influenza H7N9 Genotypes in China. Cell Host Microbe 2013, 14, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Li, K.S.; Guan, Y.; Wang, J.; Smith, G.J.D.; Xu, K.M.; Duan, L.; Rahardjo, A.P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nat. Cell Biol. 2004, 430, 209–213. [Google Scholar] [CrossRef]
- Bui, C.; Bethmont, A.; Chughtai, A.A.; Gardner, L.; Sarkar, S.; Hassan, S.; Seale, H.; MacIntyre, C.R. A Systematic Review of the Comparative Epidemiology of Avian and Human Influenza A H5N1 and H7N9—Lessons and Unanswered Questions. Transbound. Emerg. Dis. 2015, 63, 602–620. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.S.; Dusek, R.J.; Spackman, E. Rapidly Expanding Range of Highly Pathogenic Avian Influenza Viruses. Emerg. Infect. Dis. 2015, 21, 1251–1252. [Google Scholar] [CrossRef]
- Diskin, E.R.; Friedman, K.; Krauss, S.; Nolting, J.M.; Poulson, R.L.; Slemons, R.D.; Stallknecht, D.E.; Webster, R.G.; Bowman, A.S. Subtype Diversity of Influenza A Virus in North American Waterfowl: A Multidecade Study. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Deliberto, T.J.; Swafford, S.R.; Nolte, D.L.; Pedersen, K.; Lutman, M.W.; Schmit, B.B.; Baroch, J.A.; Kohler, D.J.; Franklin, A. Surveillance for highly pathogenic avian influenza in wild birds in the USA. Integr Zool 2009, 4, 426–439. [Google Scholar] [CrossRef] [Green Version]
- Szretter, K.J.; Balish, A.L.; Katz, J.M. Influenza: Propagation, quantification, and storage. Curr. Protoc. Microbiol. 2006, 3, 15G-1. [Google Scholar] [CrossRef]
- Zhou, B.; Wentworth, D.E. Influenza A Virus Molecular Virology Techniques. Advanced Struct. Saf. Stud. 2012, 865, 175–192. [Google Scholar] [CrossRef]
- Shepard, S.S.; Meno, S.; Bahl, J.; Wilson, M.M.; Barnes, J.; Neuhaus, E. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genom. 2016, 17, 708. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, H.J.; Blitvich, B.J.; VanDalen, K.; Bentler, K.T.; Franklin, A.B.; Root, J.J. Evaluation of an epitope-blocking enzyme-linked immunosorbent assay for the detection of antibodies to influenza A virus in domestic and wild avian and mammalian species. J. Virol. Methods 2009, 161, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Van, R.G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 1996. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Oliphant, T.E.; Millma, J.K. A Guide to NumPy; Trelgol Publishing: USA, 2006; Volume 1. [Google Scholar]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and statistical modeling with python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28–30 June 2010. [Google Scholar]
- Spellerberg, I.F.; Fedor, P.J. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon-Wiener’ Index. Glob. Ecol. Biogeogr. 2003, 12, 177–179. [Google Scholar] [CrossRef] [Green Version]
- Grünwald, N.J.; Goodwin, S.B.; Milgroom, M.G.; Fry, W.E. Analysis of Genotypic Diversity Data for Populations of Microorganisms. Phytopathology 2003, 93, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Mueller, J.C. Linkage disequilibrium for different scales and applications. Brief. Bioinform. 2004, 5, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, M.; Tolf, C.; Avril, A.; Latorre-Margalef, N.; Wallerström, S.; Olsen, B.; Waldenström, J. Frequency and patterns of reassortment in natural influenza A virus infection in a reservoir host. Virology 2013, 443, 150–160. [Google Scholar] [CrossRef]
- Deng, G.; Tan, D.; Shi, J.; Cui, P.; Jiang, Y.; Liu, L.; Tian, G.; Kawaoka, Y.; Li, C.; Chen, H. Complex Reassortment of Multiple Subtypes of Avian Influenza Viruses in Domestic Ducks at the Dongting Lake Region of China. J. Virol. 2013, 87, 9452–9462. [Google Scholar] [CrossRef] [Green Version]
- Dugan, V.G.; Chen, R.; Spiro, D.J.; Sengamalay, N.; Zaborsky, J.; Ghedin, E.; Nolting, J.; Swayne, D.E.; Runstadler, J.A.; Happ, G.M.; et al. The Evolutionary Genetics and Emergence of Avian Influenza Viruses in Wild Birds. PLoS Pathog. 2008, 4, e1000076. [Google Scholar] [CrossRef] [Green Version]
- Hatchette, T.F.; Walker, D.; Johnson, C.; Baker, A.; Pryor, S.P.; Webster, R.G. Influenza A viruses in feral Canadian ducks: Extensive reassortment in nature. J. Gen. Virol. 2004, 85, 2327–2337. [Google Scholar] [CrossRef]
- Macken, C.A.; Webby, R.J.; Bruno, W.J. Genotype turnover by reassortment of replication complex genes from avian Influenza A virus. J. Gen. Virol. 2006, 87, 2803–2815. [Google Scholar] [CrossRef]
- Venkatesh, D.; Poen, M.J.; Bestebroer, T.M.; Scheuer, R.D.; Vuong, O.; Chkhaidze, M.; Machablishvili, A.; Mamuchadze, J.; Ninua, L.; Fedorova, N.B.; et al. Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, B.; Ma, S.; Cui, P.; Liu, W.; Li, Y.; Guo, J.; Chen, H. High frequency of reassortment after co-infection of chickens with the H4N6 and H9N2 influenza A viruses and the biological characteristics of the reassortants. Vet. Microbiol. 2018, 222, 11–17. [Google Scholar] [CrossRef]
- Lam, T.T.-Y.; Ip, H.S.; Ghedin, E.; Wentworth, D.E.; Halpin, R.A.; Stockwell, T.B.; Spiro, D.J.; Dusek, R.J.; Bortner, J.B.; Hoskins, J.; et al. Migratory flyway and geographical distance are barriers to the gene flow of influenza virus among North American birds. Ecol. Lett. 2011, 15, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Krauss, S.; Walker, D.; Pryor, S.P.; Niles, L.; Cheng, H.L.; Hinshaw, V.S.; Webster, R.G. Influenza A viruses of migrating wild aquatic birds in North America. Vector-Borne Zoonotic Dis. 2004, 4, 177–189. [Google Scholar] [CrossRef]
- Hill, N.J.; Ma, E.J.; Meixell, B.W.; Lindberg, M.S.; Boyce, W.M.; Runstadler, J.A. Transmission of influenza reflects seasonality of wild birds across the annual cycle. Ecol. Lett. 2016, 19, 915–925. [Google Scholar] [CrossRef]
- McCrone, J.T.; Woods, R.J.; Martin, E.T.; E Malosh, R.; Monto, A.S.; Lauring, A.S. Stochastic processes constrain the within and between host evolution of influenza virus. eLife 2018, 7, e35962. [Google Scholar] [CrossRef] [PubMed]
- Varble, A.; Albrecht, R.A.; Backes, S.; Crumiller, M.; Bouvier, N.M.; Sachs, D.; García-Sastre, A.; Tenoever, B.R. Influenza A Virus Transmission Bottlenecks Are Defined by Infection Route and Recipient Host. Cell Host Microbe 2014, 16, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Lowen, A.C. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Annu. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence |
|---|---|
| PB2 F | CATGCTGGGAGCAAATGTACA |
| PB2 R | TCTTACTATGTTCCTGGCAGC |
| PB1 F | GGAACAGGATACACCATGGA |
| PB1 R | GTTGTCCATTTCCCCTTTTCTG |
| PA F | GGGATTCCTTTCGTCAGTC |
| PA R | GCCTGCGCATGGTTC |
| NP F | AGGGCACTTGTGCGTACT |
| NP R | CCTTTCACTGCTGCTCCA |
| M F | GCATCGGTCTCACAGACA |
| M R | CCTGCCATTTGCTCCATG |
| NS F | GAATCCGACGAGGCACT |
| NS R | TGGGCATGAGCATGAACC |
| H3 F | AAATGGAGGGAGTGGAGCTT |
| H3 R | TGAACTCCCCACACGTACAA |
| H4 F | CTGCCCAGGAATTAGTGGAA |
| H4 R | TCTGGCACATCAAATGGGTA |
| N8 F | TGAAAGACCGGAGCCCCTAT |
| N8 R | AGGGCCCGTTACTCCAATTG |
| N6 F | TTGGGAAATGGGGCAAGCA |
| N6 R | GCCTTCCCTTGTACCAGACC |
| Segment | % Nucleotide Identity | Protein | % Amino Acid Identity |
|---|---|---|---|
| PB2 | 91.3 | PB2 | 98.8 |
| PB1 | 94.3 | PB1 PB1-F2 | 99.2 85.6 |
| PA | 87.7 | PA PA-X | 98.3 98.4 |
| HA | 65.0 | HA | 68.7 |
| NP | 93.7 | NP | 100 |
| NA | 55.6 | NA | 44.2 |
| M | 98.2 | M1 M2 | 100 99.0 |
| NS | 94.6 | NS1 NEP | 98.7 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganti, K.; Bagga, A.; DaSilva, J.; Shepard, S.S.; Barnes, J.R.; Shriner, S.; Koelle, K.; Lowen, A.C. Avian Influenza A Viruses Reassort and Diversify Differently in Mallards and Mammals. Viruses 2021, 13, 509. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030509
Ganti K, Bagga A, DaSilva J, Shepard SS, Barnes JR, Shriner S, Koelle K, Lowen AC. Avian Influenza A Viruses Reassort and Diversify Differently in Mallards and Mammals. Viruses. 2021; 13(3):509. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030509
Chicago/Turabian StyleGanti, Ketaki, Anish Bagga, Juliana DaSilva, Samuel S. Shepard, John R. Barnes, Susan Shriner, Katia Koelle, and Anice C. Lowen. 2021. "Avian Influenza A Viruses Reassort and Diversify Differently in Mallards and Mammals" Viruses 13, no. 3: 509. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030509