
Conserved Structural Motifs of Two Distant IAV Subtypes in Genomic Segment 5 RNA

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Oligonucleotide Synthesis and Labeling
2.2. Synthesis of DNA Template of vRNA5 A/California/04/2009
2.3. Transcription In Vitro
2.4. Chemical Mapping
2.5. Reverse Transcription and Primer Extension
2.6. Data analysis
2.6.1. Primer-Extension Data Analysis
2.6.2. Secondary Structure Prediction and Base-Pairing Probability Calculation
2.6.3. Conservation of Canonical Base-Pairing Calculation
3. Results and Discussion
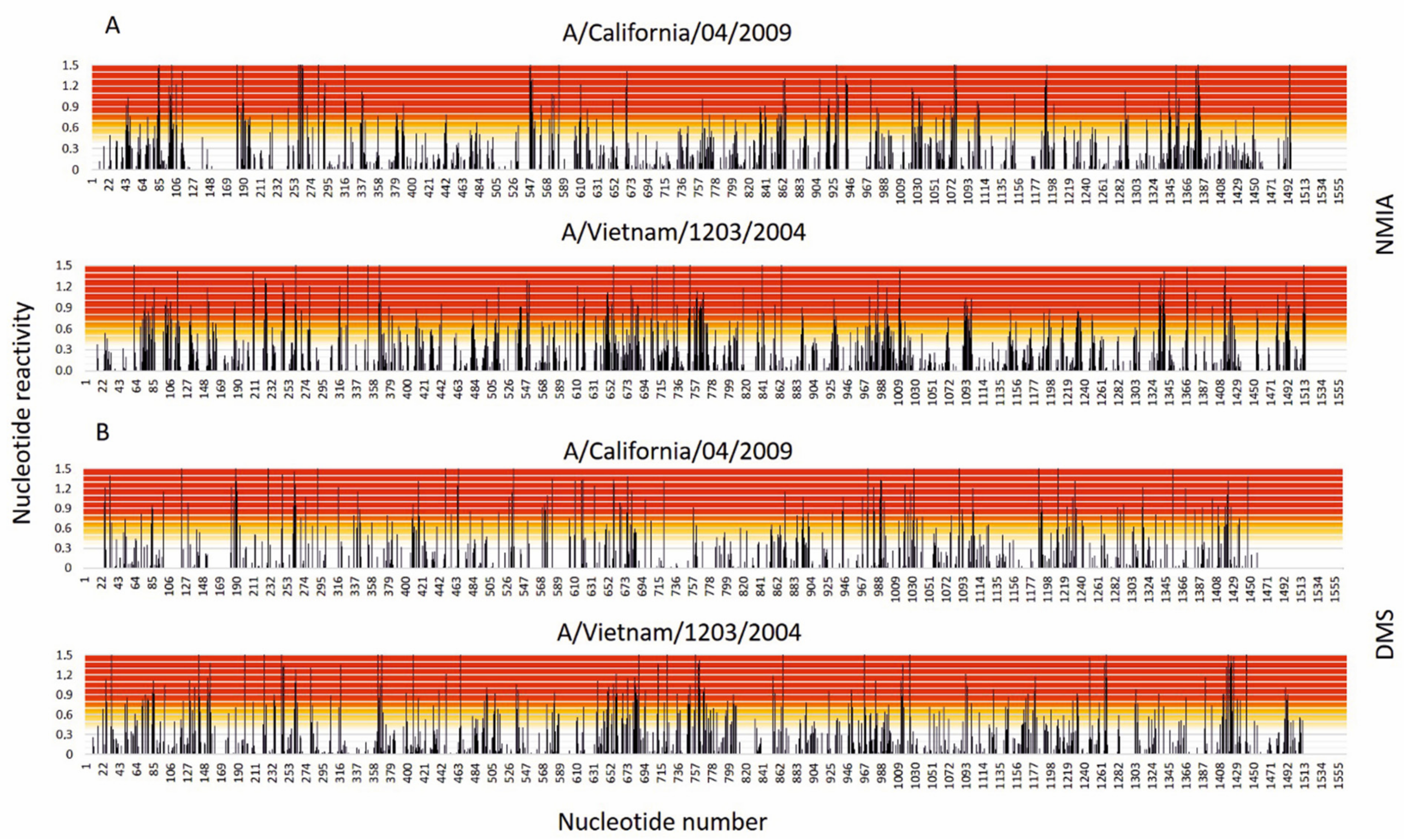
3.1. Results of Chemical Mapping
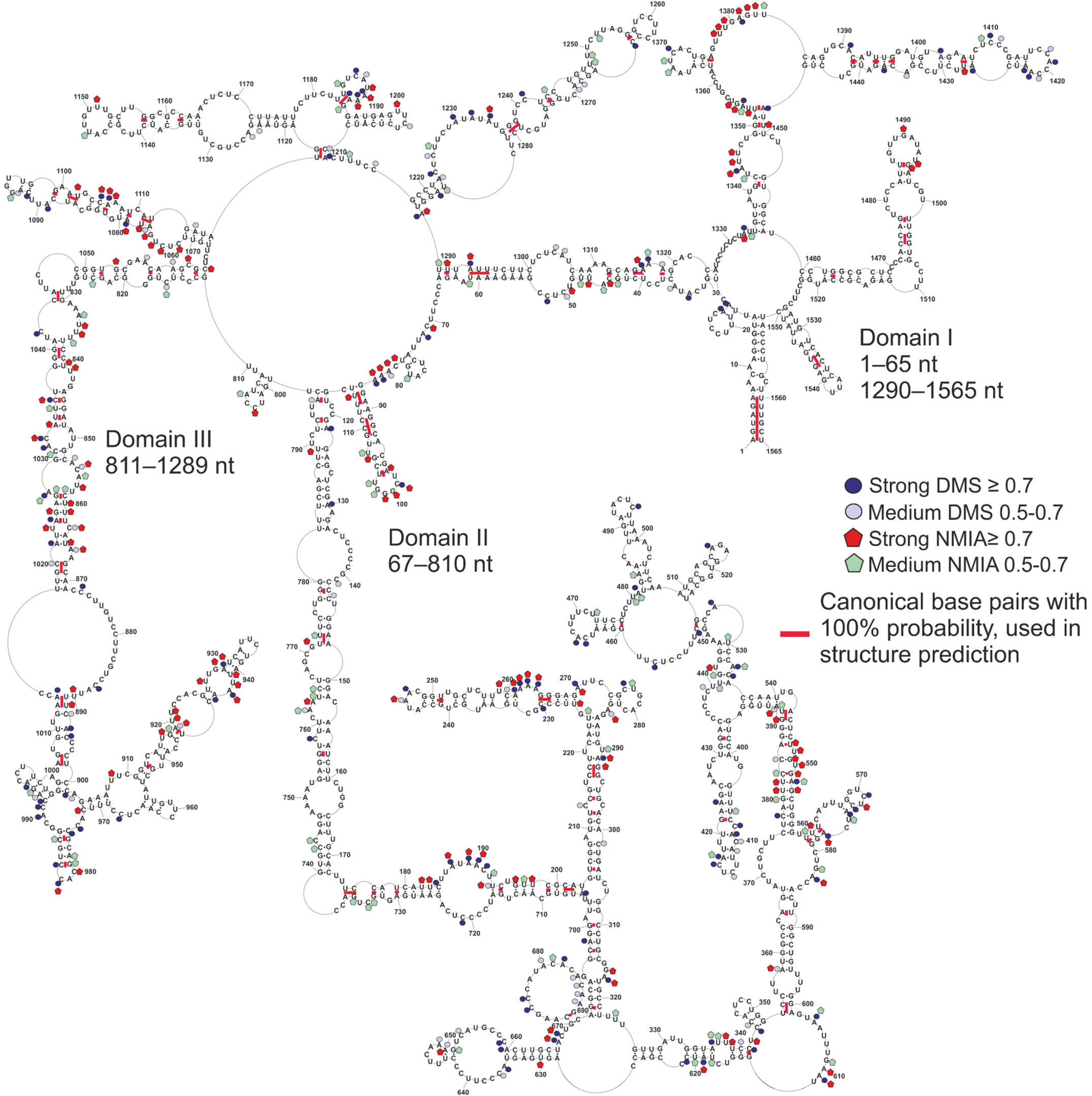
3.2. Global Folding of vRNA5 A/California/04/2009
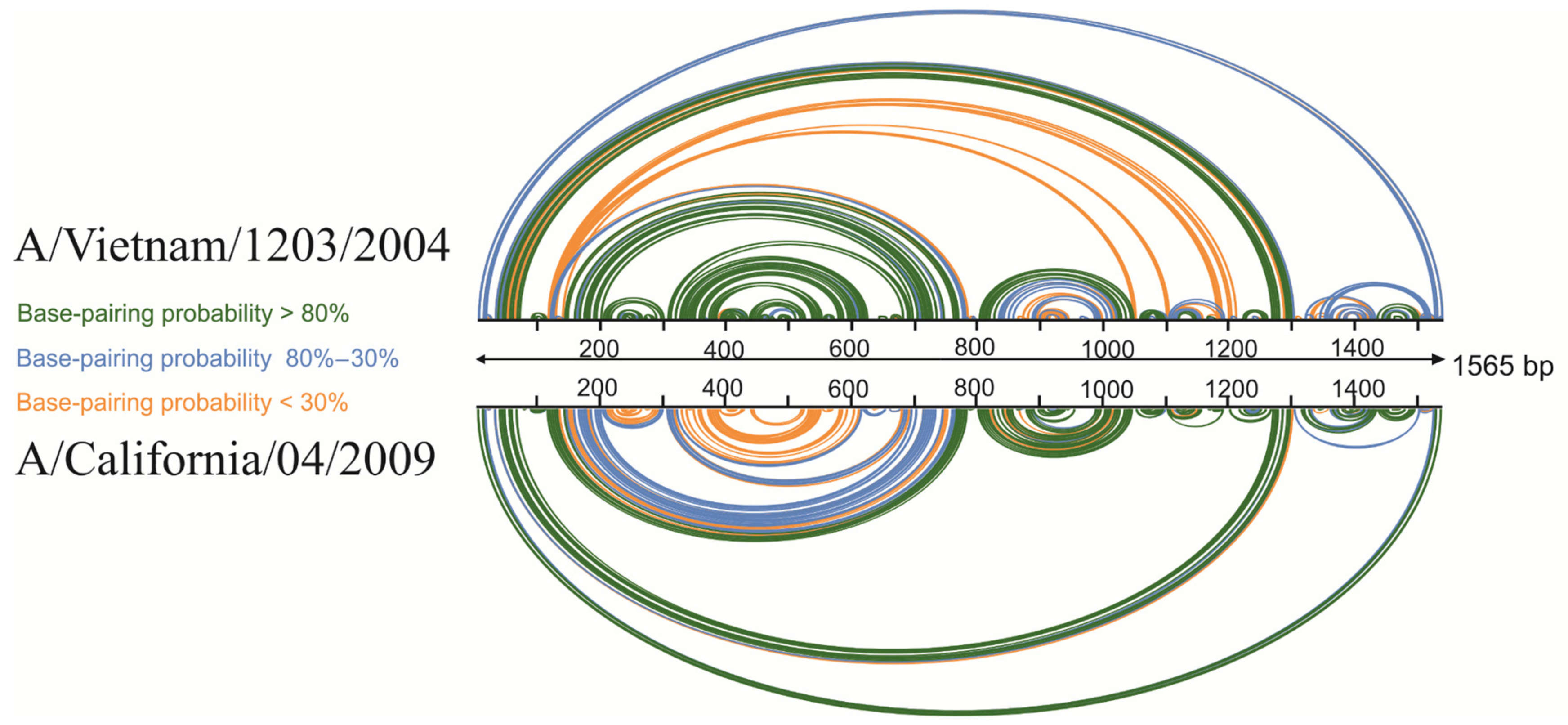
3.3. Base-Pairing Probabilities Based on Experimental Data
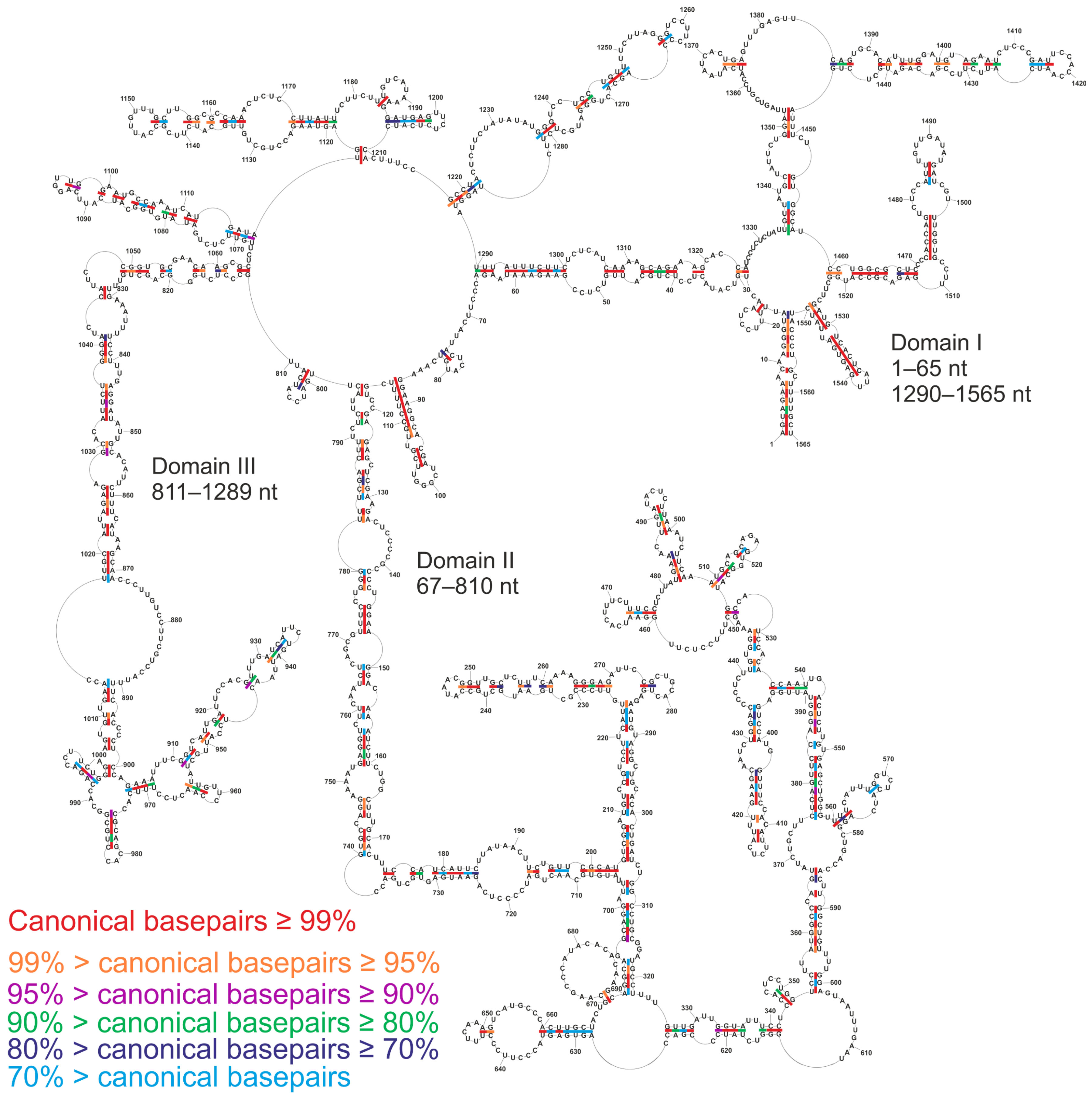
3.4. Conservation of Canonical Base-Pairing in vRNA5
3.5. Secondary Structure of vRNA5 and Antisense Oligonucleotides (ASOs) Inhibitory Potential
3.6. Mirror Structures in vRNA5 within Distant IAV Strains
3.7. NP-Binding Profile In Vivo Influences ASOs Inhibitory Potential
3.8. The Structure of the vRNP5 Complex Revealed Interacting Regions
3.9. The Secondary Structure of vRNA5 within Different IAV Strains
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mostafa, A.; Abdelwhab, E.M.; Mettenleiter, T.C.; Pleschka, S. Zoonotic potential of influenza A viruses: A comprehensive overview. Viruses 2018, 10, 497. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, S.; Kawaoka, Y. The pathogenesis of influenza virus infections: The contributions of virus and host factors. Curr. Opin. Immunol. 2011, 23, 481–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amesh, A.A.; Matthew, W.; Toner, E.S.; Cicero, A.; Inglesby, T.V. Characteristics of Microbes Most Likely to Cause Pandemics and Global Catastrophes; Springer Nature: Cham, Switzerland, 2019; Volume 424. [Google Scholar] [CrossRef]
- Epidemic and Pandemic Alert Response Unit. WHO Guidelines for Investigation of Human Cases of Avian Influenza A(H5N1); World Health Organization: Geneva, Switzerland, 2007. [Google Scholar]
- Li, K.S.; Guan, Y.; Wang, J.; Smith, G.J.D.; Xu, K.M.; Duan, L.; Rahardjo, A.P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 2004, 430, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Gultyaev, A.P.; Fouchier, R.A.M.; Olsthoorn, R.C.L. Influenza virus RNA structure: Unique and common features. Int. Rev. Immunol. 2010, 29, 533–556. [Google Scholar] [CrossRef]
- Ferhadian, D.; Contrant, M.; Printz-Schweigert, A.; Smyth, R.P.; Paillart, J.C.; Marquet, R. Structural and functional motifs in influenza virus RNAs. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wandzik, J.M.; Kouba, T.; Karuppasamy, M.; Pflug, A.; Drncova, P.; Provaznik, J.; Azevedo, N.; Cusack, S. A structure-based model for the complete transcription cycle of influenza polymerase. Cell 2020, 181, 877–893. [Google Scholar] [CrossRef]
- Ruszkowska, A.; Lenartowicz, E.; Moss, W.N.; Kierzek, R.; Kierzek, E. Secondary structure model of the naked segment 7 influenza A virus genomic RNA. Biochem. J. 2016, 473, 4327–4348. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, E.; Kesy, J.; Ruszkowska, A.; Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.; Turner, D.H.; Kierzek, R.; Kierzek, E. Self-folding of naked segment 8 genomic RNA of influenza a virus. PLoS ONE 2016, 11, e0148281. [Google Scholar] [CrossRef] [Green Version]
- Michalak, P.; Soszynska-Jozwiak, M.; Biala, E.; Moss, W.N.; Kesy, J.; Szutkowska, B.; Lenartowicz, E.; Kierzek, R.; Kierzek, E. Secondary structure of the segment 5 genomic RNA of influenza A virus and its application for designing antisense oligonucleotides. Sci. Rep. 2019, 9, 3801. [Google Scholar] [CrossRef] [Green Version]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat. Microbiol. 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Le Sage, V.; Kanarek, J.P.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S.; Lee, N. Mapping of influenza virus RNA-RNA interactions reveals a flexible network. Cell Rep. 2020, 31, 107823. [Google Scholar] [CrossRef]
- Takizawa, N.; Higashi, K.; Kawaguchi, R.K.; Gotoh, Y.; Suzuki, Y.; Hayashi, T.; Kurokawa, K. A functional RNA structure in the influenza A virus ribonucleoprotein complex for segment bundling. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, S.J. Challenges and approaches to predicting RNA with multiple functional structures. RNA 2018, 24, 1615–1624. [Google Scholar] [CrossRef] [Green Version]
- Gavazzi, C.; Yver, M.; Isel, C.; Smyth, R.P.; Rosa-Calatrava, M.; Lina, B.; Moulès, V.; Marquet, R. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc. Natl. Acad. Sci. USA 2013, 110, 16604–16609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Dadonaite, B.; van Doremalen, N.; Suzuki, Y.; Barclay, W.S.; Pybus, O.G. Computational and molecular analysis of conserved influenza A virus RNA secondary structures involved in infectious virion production. RNA Biol. 2016, 13, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.D.; Townsend, D.; Wylie, K.M.; Kim, P.J.; Amarasinghe, G.K.; Kutluay, S.B.; Boon, A.C.M. Nucleotide resolution mapping of influenza A virus nucleoprotein-RNA interactions reveals RNA features required for replication. Nat. Commun. 2018, 9, 465. [Google Scholar] [CrossRef] [Green Version]
- Gultyaev, A.P.; Tsyganov-Bodounov, A.; Spronken, M.I.J.; Van Der Kooij, S.; Fouchier, R.A.M.; Olsthoorn, R.C.L. RNA structural constraints in the evolution of the influenza A virus genome NP segment. RNA Biol. 2014, 11, 942–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, J.; Jarmolowicz, A.; Kierzek, E. Organization of the Influenza A Virus Genomic RNA in the Viral Replication Cycle—Structure, Interactions, and Implications for the Emergence of New Strains. Pathogens 2020, 9, 951. [Google Scholar] [CrossRef] [PubMed]
- Gultyaev, A.P.; Spronken, M.I.; Richard, M.; Schrauwen, E.J.A.; Olsthoorn, R.C.L.; Fouchier, R.A.M. Subtype-specific structural constraints in the evolution of influenza A virus hemagglutinin genes. Sci. Rep. 2016, 6, 38892. [Google Scholar] [CrossRef] [Green Version]
- Spronken, M.I.; van de Sandt, C.E.; de Jongh, E.P.; Vuong, O.; van der Vliet, S.; Bestebroer, T.M.; Olsthoorn, R.C.L.; Rimmelzwaan, G.F.; Fouchier, R.A.M.; Gultyaev, A.P. A compensatory mutagenesis study of a conserved hairpin in the M gene segment of influenza A virus shows its role in virus replication. RNA Biol. 2017, 14, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Bolte, H.; Rosu, M.E.; Hagelauer, E.; García-Sastre, A.; Schwemmle, M. Packaging of the influenza virus genome is governed by a plastic network of RNA- and nucleoprotein-mediated interactions. J. Virol. 2018, 93, e01861-18. [Google Scholar] [CrossRef] [Green Version]
- Triggle, C.R.; Bansal, D.; Farag Elmoubasher, A.B.A.; Hong, D. COVID-19: Learning from Lessons to Guide Treatment and Prevention Interventions. Am. Soc. Microbiol. 2020, 5, 1–13. [Google Scholar]
- Duwe, S. Influenza viruses—Antiviral therapy and resistance. GMS Infect. Dis. 2017, 5. [Google Scholar] [CrossRef]
- Saito, R.; Sakai, T.; Sato, I.; Sano, Y.; Oshitani, H.; Sato, M.; Suzuki, H. Frequency of amantadine-resistant influenza A viruses during two seasons featuring cocirculation of H1N1 and H3N2. J. Clin. Microbiol. 2003, 41, 2164–2165. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Young, J.S.; Jeung, H.P.; Lee, J.H.; Yun, H.B.; Song, M.S.; Oh, T.K.; Han, H.S.; Pascua, P.N.Q.; Choi, Y.K. Emergence of amantadine-resistant H3N2 avian influenza A virus in South Korea. J. Clin. Microbiol. 2008, 46, 3788–3790. [Google Scholar] [CrossRef] [Green Version]
- Zaraket, H.; Saito, R.; Suzuki, Y.; Baranovich, T.; Dapat, C.; Caperig-Dapat, I.; Suzuki, H. Genetic makeup of amantadine-resistant and oseltamivir-resistant human influenza A/H1N1 viruses. J. Clin. Microbiol. 2010, 48, 1085–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piasecka, J.; Lenartowicz, E.; Soszynska-Jozwiak, M.; Szutkowska, B.; Kierzek, R.; Kierzek, E. RNA secondary structure motifs of the influenza A virus as targets for siRNA-mediated RNA interference. Mol. Ther. Nucleic Acids 2020, 19, 627–642. [Google Scholar] [CrossRef]
- Lenartowicz, E.; Nogales, A.; Kierzek, E.; Kierzek, R.; Martínez-Sobrido, L.; Turner, D.H. Antisense oligonucleotides targeting influenza A segment 8 genomic RNA inhibit viral replication. Nucleic Acid Ther. 2016, 26, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesy, J.; Patil, K.M.; Kumar, S.R.; Shu, Z.; Yong, H.Y.; Zimmermann, L.; Ong, A.A.L.; Toh, D.-F.K.; Krishna, M.S.; Yang, L.; et al. A short chemically modified dsRNA-binding PNA (dbPNA) inhibits influenza viral replication by targeting viral RNA panhandle structure. Bioconjug. Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef]
- Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.; Kierzek, R.; Kesy, J.; Kierzek, E. Influenza virus segment 5 (+)RNA—Secondary structure and new targets for antiviral strategies. Sci. Rep. 2017, 7, 15041. [Google Scholar] [CrossRef] [PubMed]
- Kierzek, E.; Kierzek, R. The synthesis of oligoribonucleotides containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines via post-synthetic modification of precursor oligomers. Nucleic Acids Res. 2003, 31, 4461–4471. [Google Scholar] [CrossRef] [Green Version]
- Vasa, S.M.; Guex, N.; Wilkinson, K.A.; Weeks, K.M.; Giddings, M.C. ShapeFinder: A software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA 2008, 14, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Bellaousov, S.; Reuter, J.S.; Seetin, M.G.; Mathews, D.H. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013, 41, W471–W474. [Google Scholar] [CrossRef] [Green Version]
- Mathews, D.H. Predicting a set of minimal free energy RNA secondary structures common to two sequences. Bioinformatics 2005, 21, 2246–2253. [Google Scholar] [CrossRef] [Green Version]
- Harmanci, A.O.; Sharma, G.; Mathews, D.H. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign. BMC Bioinform. 2007, 8, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The Influenza Virus Resource at the National Center for Biotechnology Information. J. Virol. 2008, 82, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA structure determination a SHAPE experiment can be interpreted as a pseudo-free energy high accuracy. Free energy minimization, by using SHAPE pseudo-. Sci. York 2008, 2008, 97–102. [Google Scholar]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [Green Version]
- Kierzek, E.; Christensen, S.M.; Eickbush, T.H.; Kierzek, R.; Turner, D.H.; Moss, W.N. Secondary Structures for 5′ Regions of R2 Retrotransposon RNAs Reveal a Novel Conserved Pseudoknot and Regions that Evolve under Different Constraints. J. Mol. Biol. 2009, 390, 428–442. [Google Scholar] [CrossRef] [Green Version]
- Gavazzi, C.; Isel, C.; Fournier, E.; Moules, V.; Cavalier, A.; Thomas, D.; Lina, B.; Marquet, R. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: Comparison with a human H3N2 virus. Nucleic Acids Res. 2013, 41, 1241–1254. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Le Sage, V.; Nanni, A.V.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res. 2017, 45, 8968–8977. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Motif Nucleotide Region (nt) | Base-Pairing Probability According to Dadonaite, et al. [16] | |
|---|---|---|
| In Virio | Ex Virio | |
| 91–111 | <10% | none |
| 406–422 | 30–80% | >80% |
| 460–476 | >80% | >80% |
| 931–941 | 30–80% | 10–30% |
| 976–986 | 30–80% | 30–80% |
| 993–1001 | >80% | <10% |
| 1194–1209 | >80% | 30–80% |
| 1363–1375 | 30–80% | 10–30% |
| 1484–1496 | 10–30% | none |
| 1527–1550 | >80% | >80% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalak, P.; Piasecka, J.; Szutkowska, B.; Kierzek, R.; Biala, E.; Moss, W.N.; Kierzek, E. Conserved Structural Motifs of Two Distant IAV Subtypes in Genomic Segment 5 RNA. Viruses 2021, 13, 525. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030525
Michalak P, Piasecka J, Szutkowska B, Kierzek R, Biala E, Moss WN, Kierzek E. Conserved Structural Motifs of Two Distant IAV Subtypes in Genomic Segment 5 RNA. Viruses. 2021; 13(3):525. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030525
Chicago/Turabian StyleMichalak, Paula, Julita Piasecka, Barbara Szutkowska, Ryszard Kierzek, Ewa Biala, Walter N. Moss, and Elzbieta Kierzek. 2021. "Conserved Structural Motifs of Two Distant IAV Subtypes in Genomic Segment 5 RNA" Viruses 13, no. 3: 525. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030525