
Role of Small Envelope Protein in Sustaining the Intracellular and Extracellular Levels of Hepatitis B Virus Large and Middle Envelope Proteins

 ,
,
Abstract
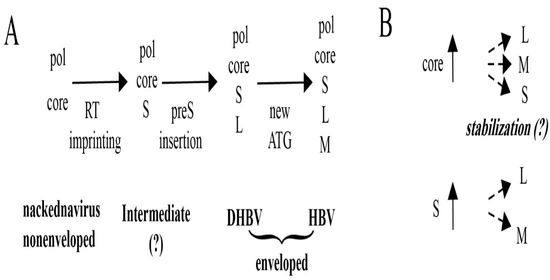
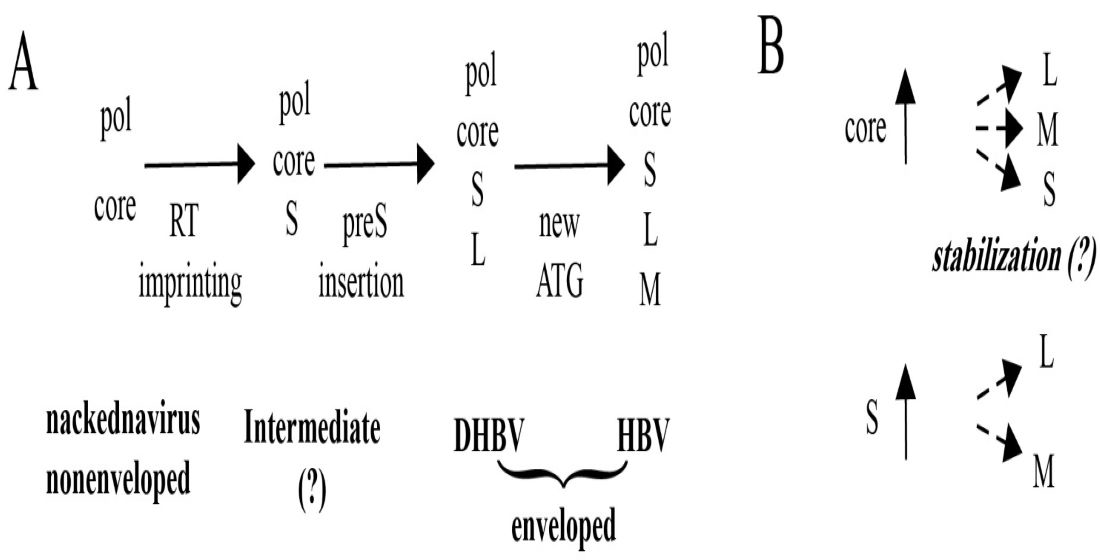
:
1. Introduction
2. Materials and Methods
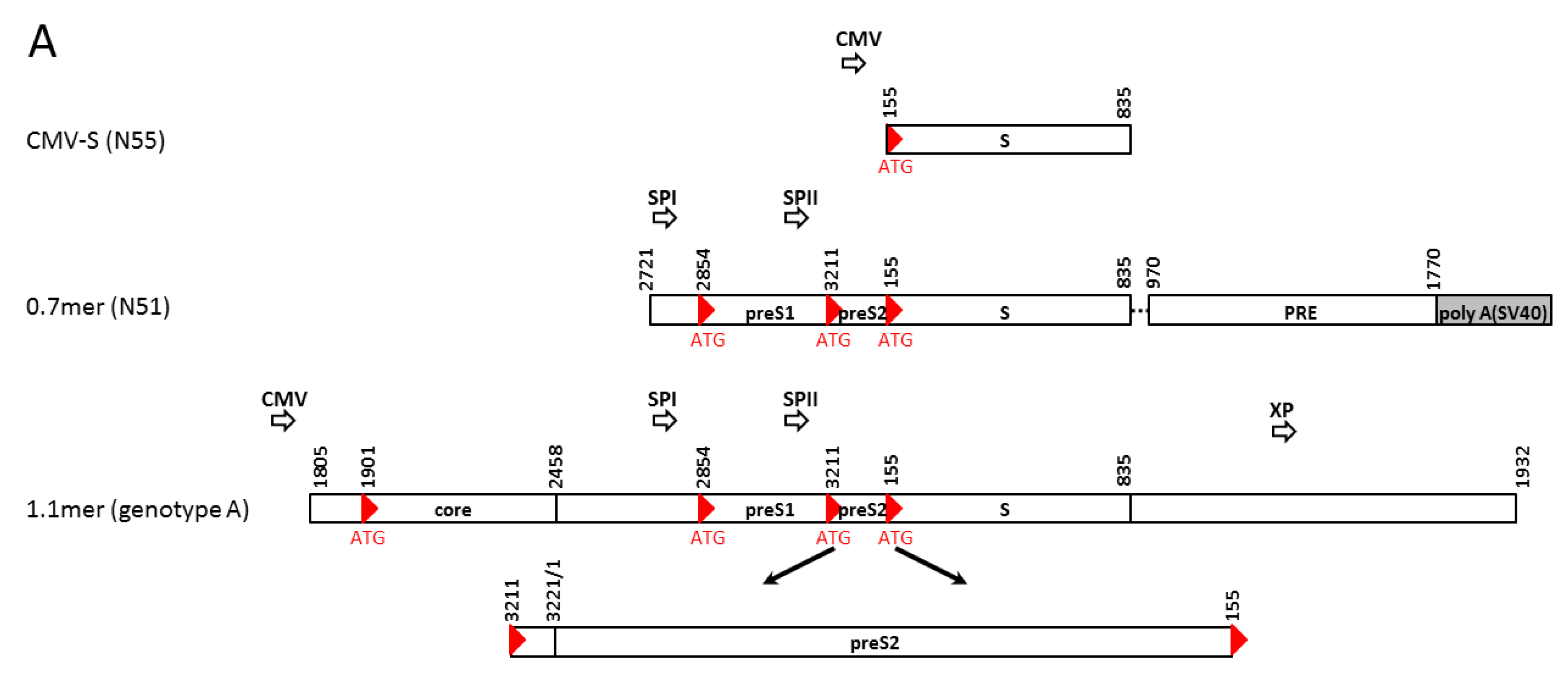
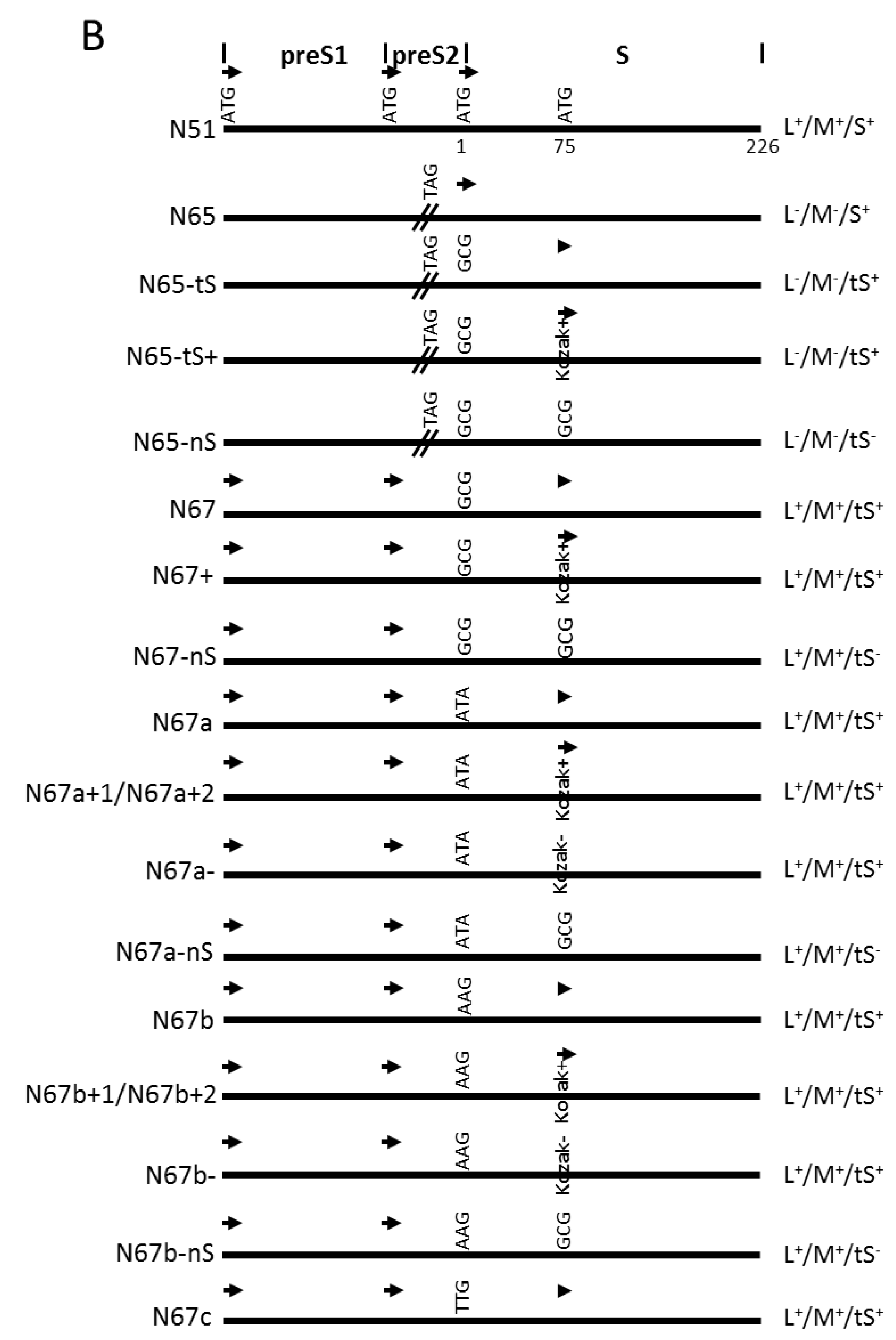
2.1. Subgenomic Expression Constructs for HBV Envelope Proteins and Site-Directed Mutants
2.2. 1.1 mer Constructs and Mutation to Prevent the Expression of S, L, or Core Protein
2.3. Transient Transfection and Western Blot Analysis
2.4. ELISA Measurement of HBsAg and preS1 Antigen
2.5. Southern Blot Analysis of Replicative DNA and Virion DNA
2.6. Statistical Analysis
3. Results
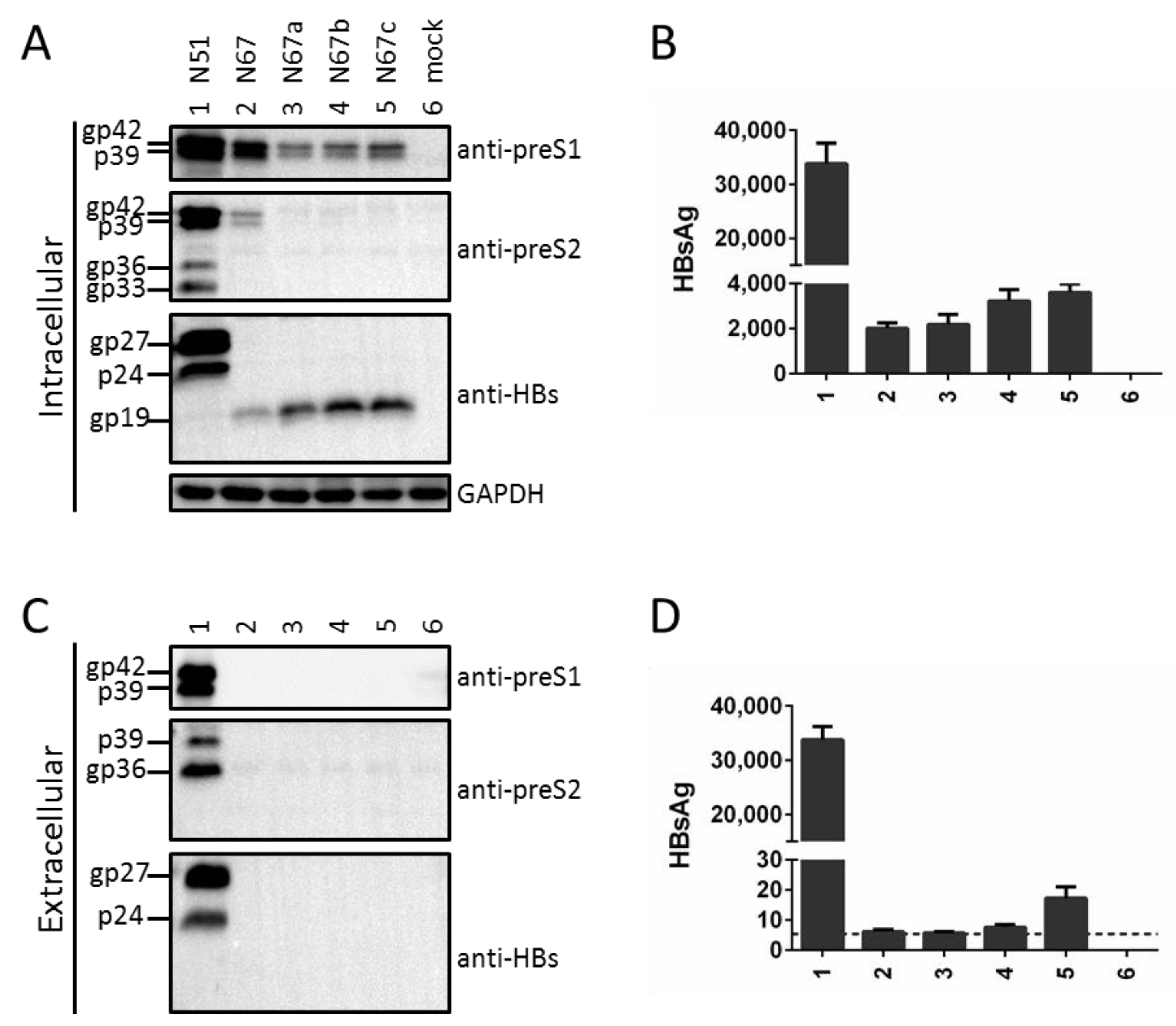
3.1. Mutating the S Gene ATG Codon from a Subgenomic L/M/S Construct of a Genotype A Clone Reduced Intracellular Level of L Protein Despite Its Blocked Secretion
3.2. Mutating the S Gene ATG Codon Led to Translation Initiation from the Next In-Frame ATG to Generate an N-Terminally Truncated S Protein
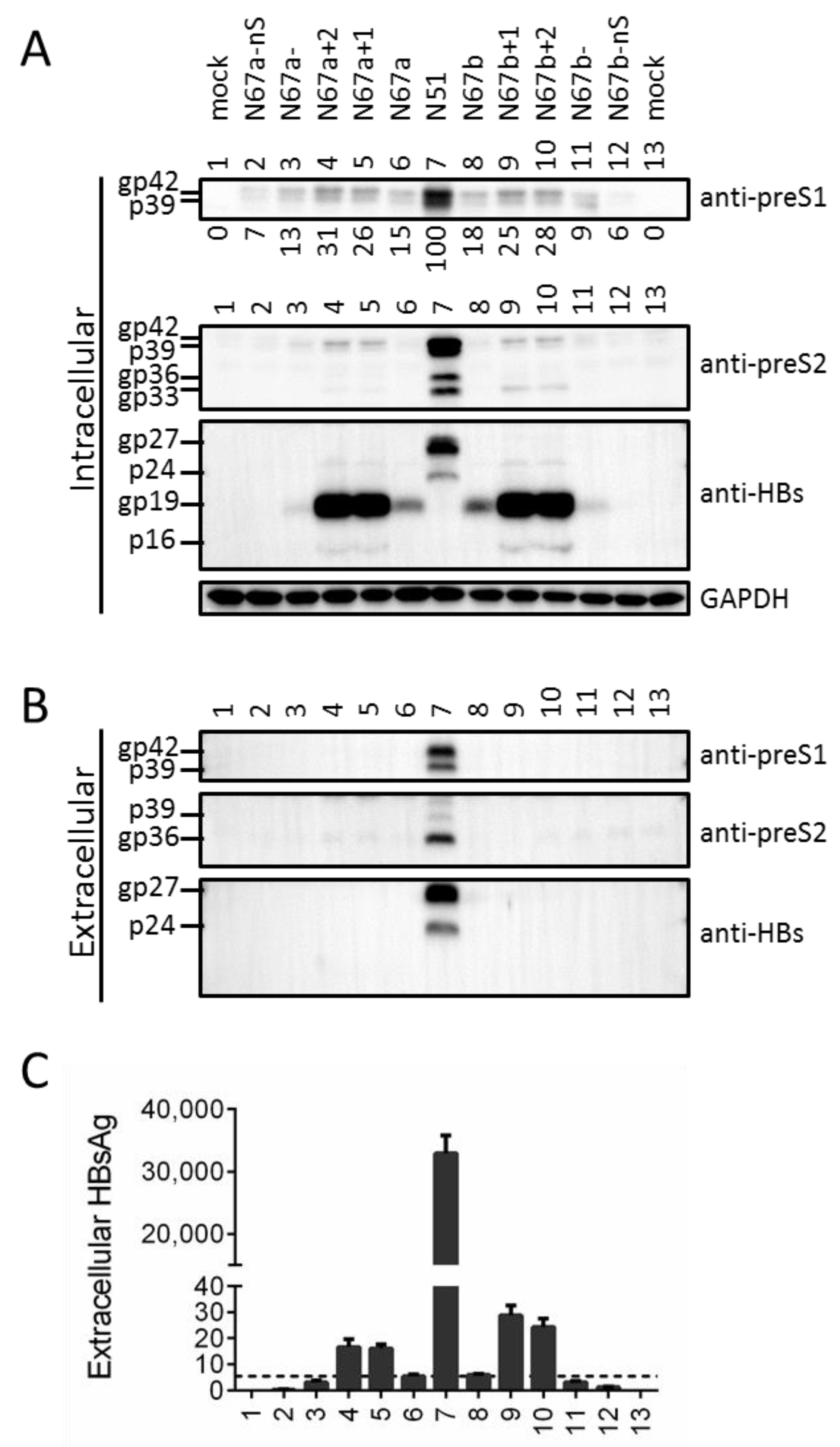
3.3. Truncated S Protein Was Secretion-Deficient but Capable of Partially Sustaining Intracellular Level of L Protein When Provided in Cis
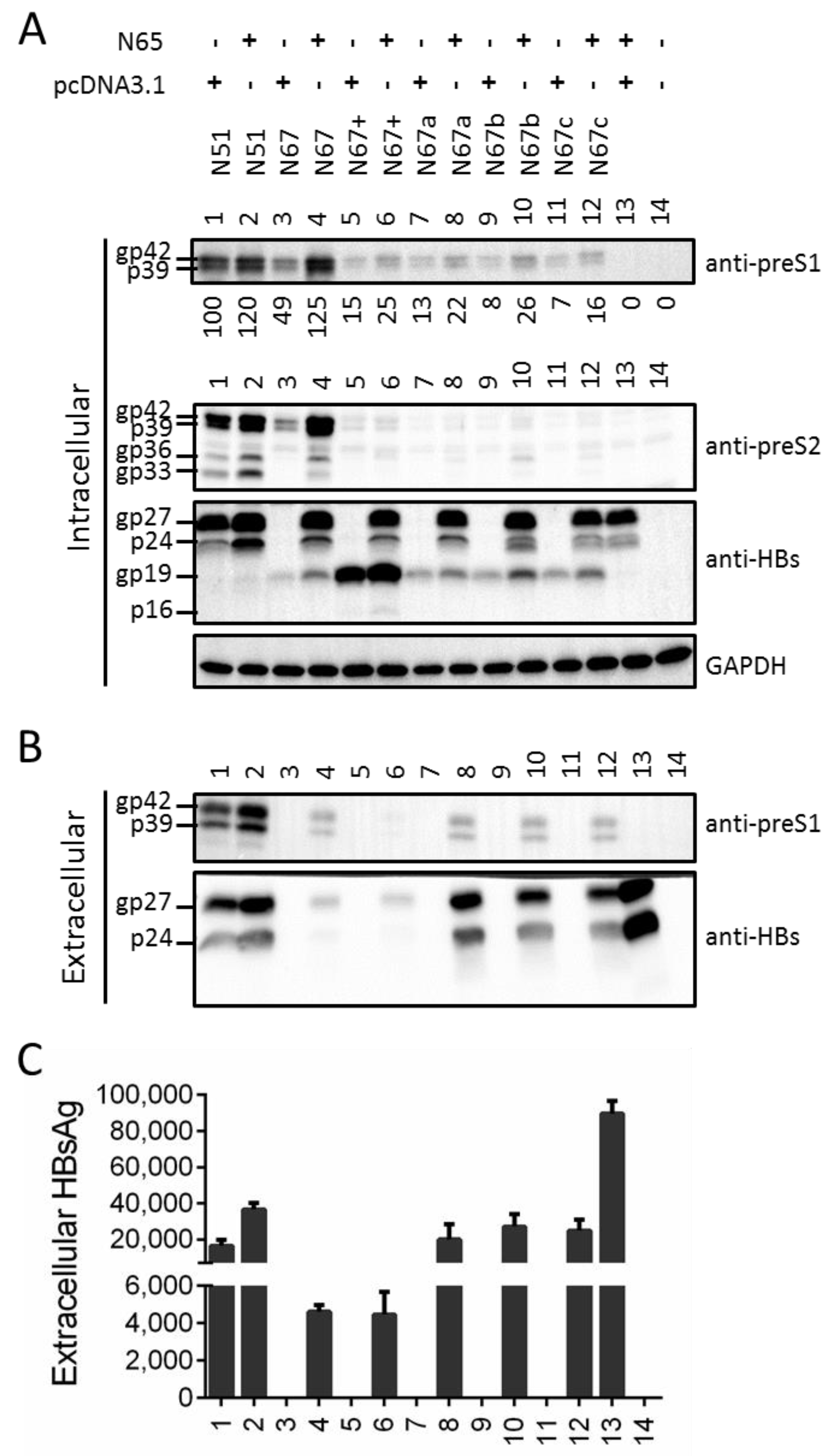
3.4. Providing Full-Length S Protein in Trans Rescued L Protein Secretion from ATG Codon Mutants and Moderately Increased Its Intracellular Level
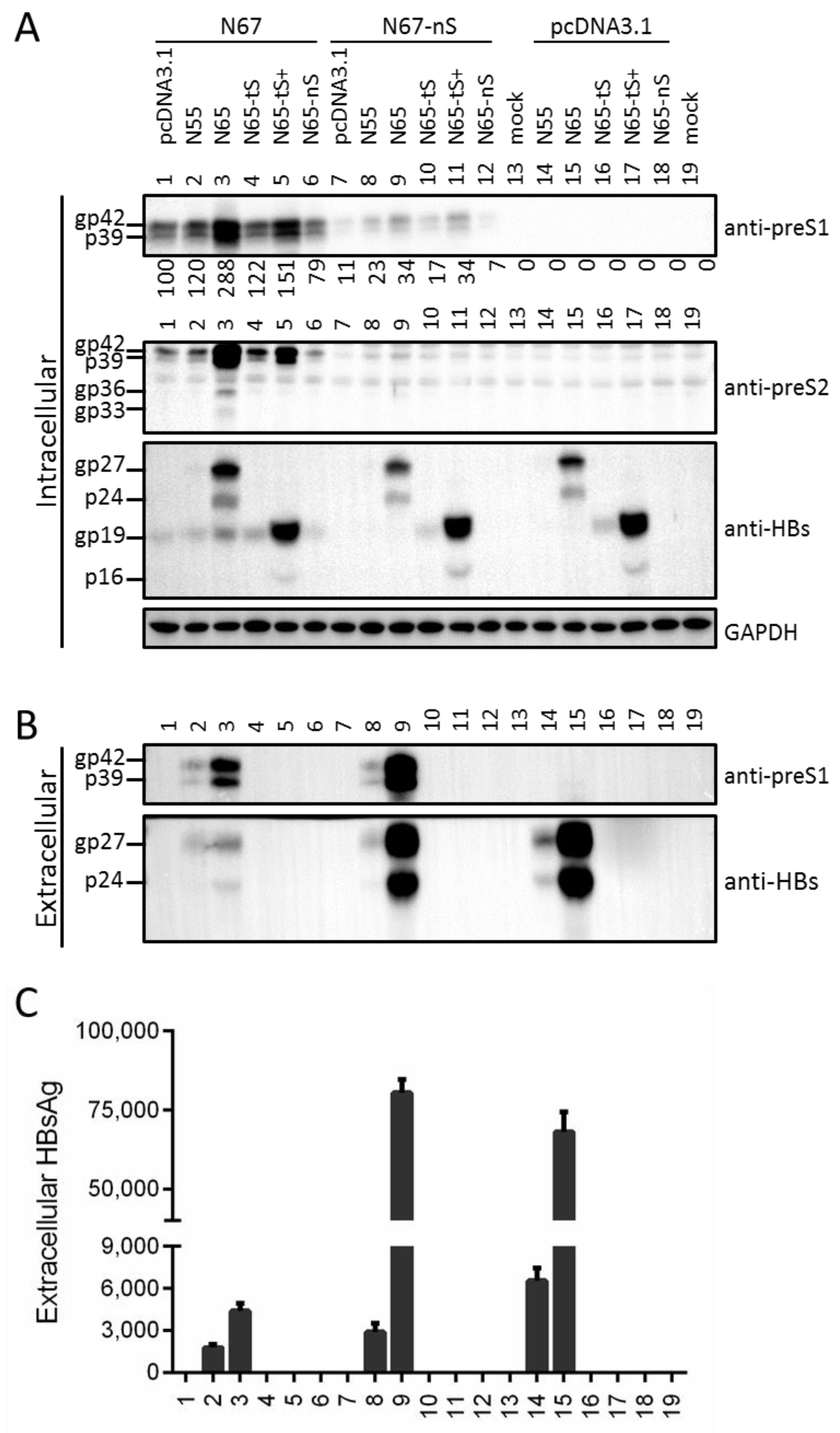
3.5. Providing Truncated S Protein in Trans Failed to Rescue L Protein Secretion from ATG Codon Mutants but Could Increase Intracellular L Protein
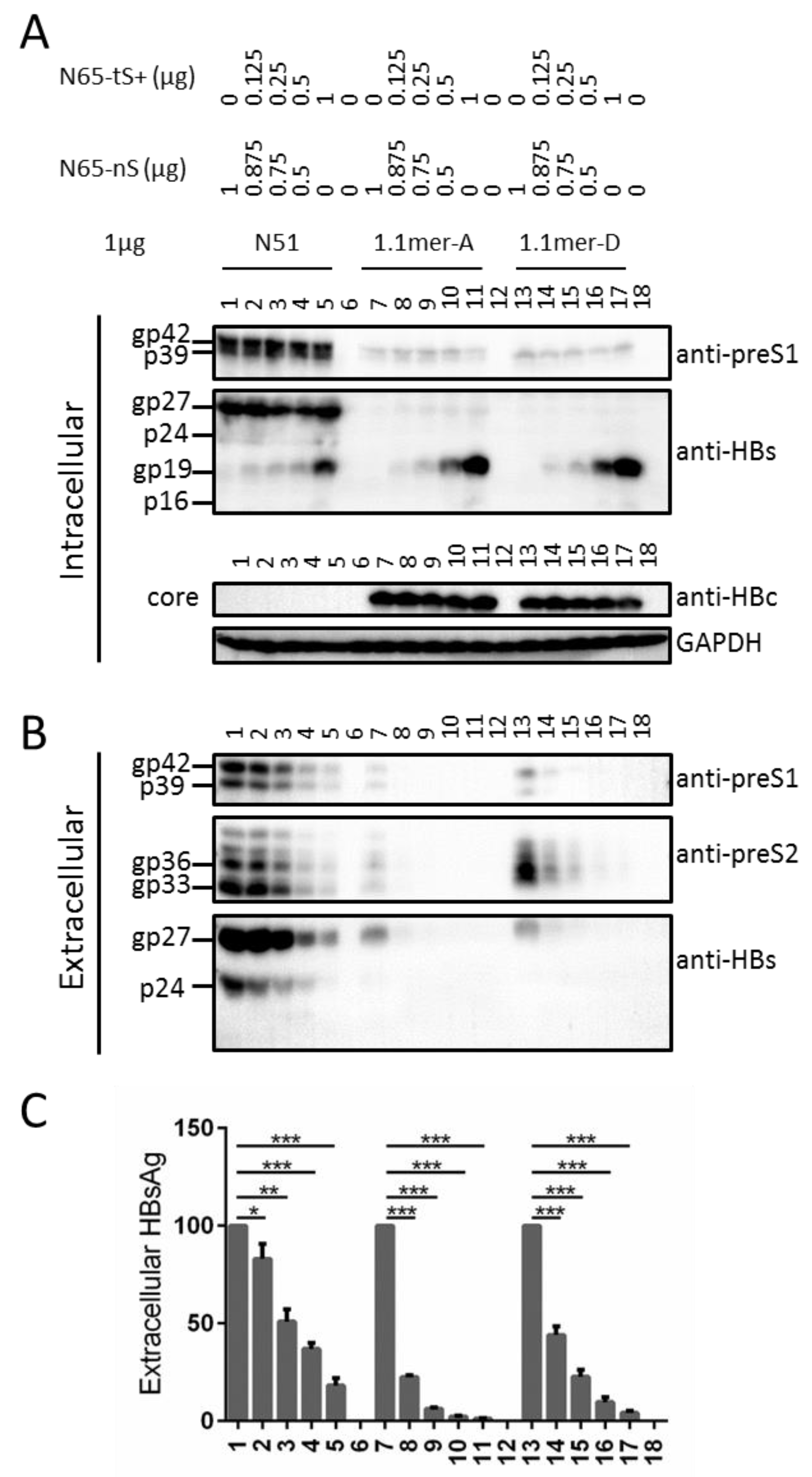
3.6. Truncated S Protein Had Dominant Negative Effect on HBsAg Secretion from Wild-Type 0.7 mer L+/M+/S+ Construct of Genotype A or 1.1 mer Construct of Genotype A or D
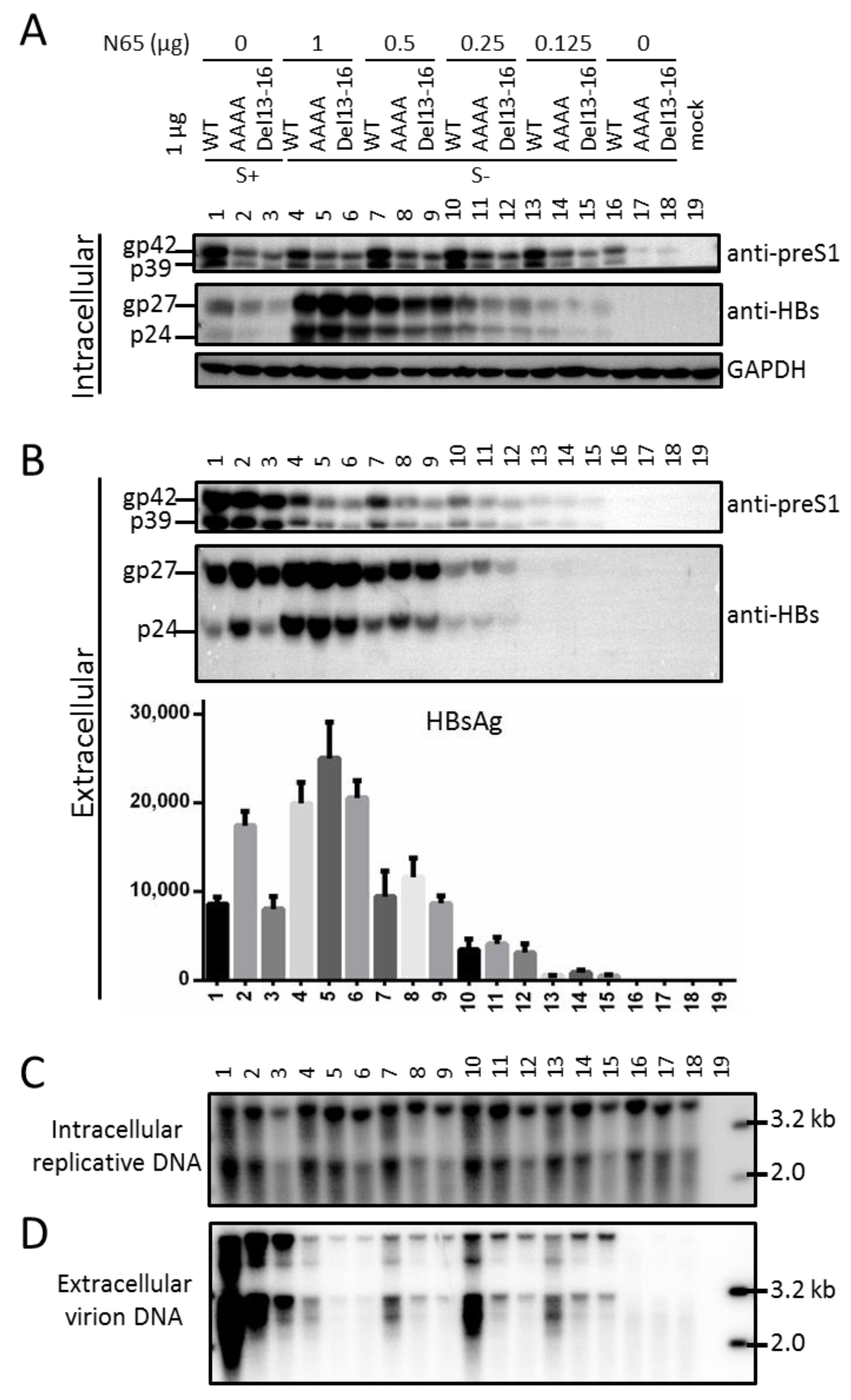
3.7. Full-Length S Protein Sustained Intracellular Level of L Protein in the Context of a 1.1 mer Construct of Genotype D with Mutated S Gene ATG
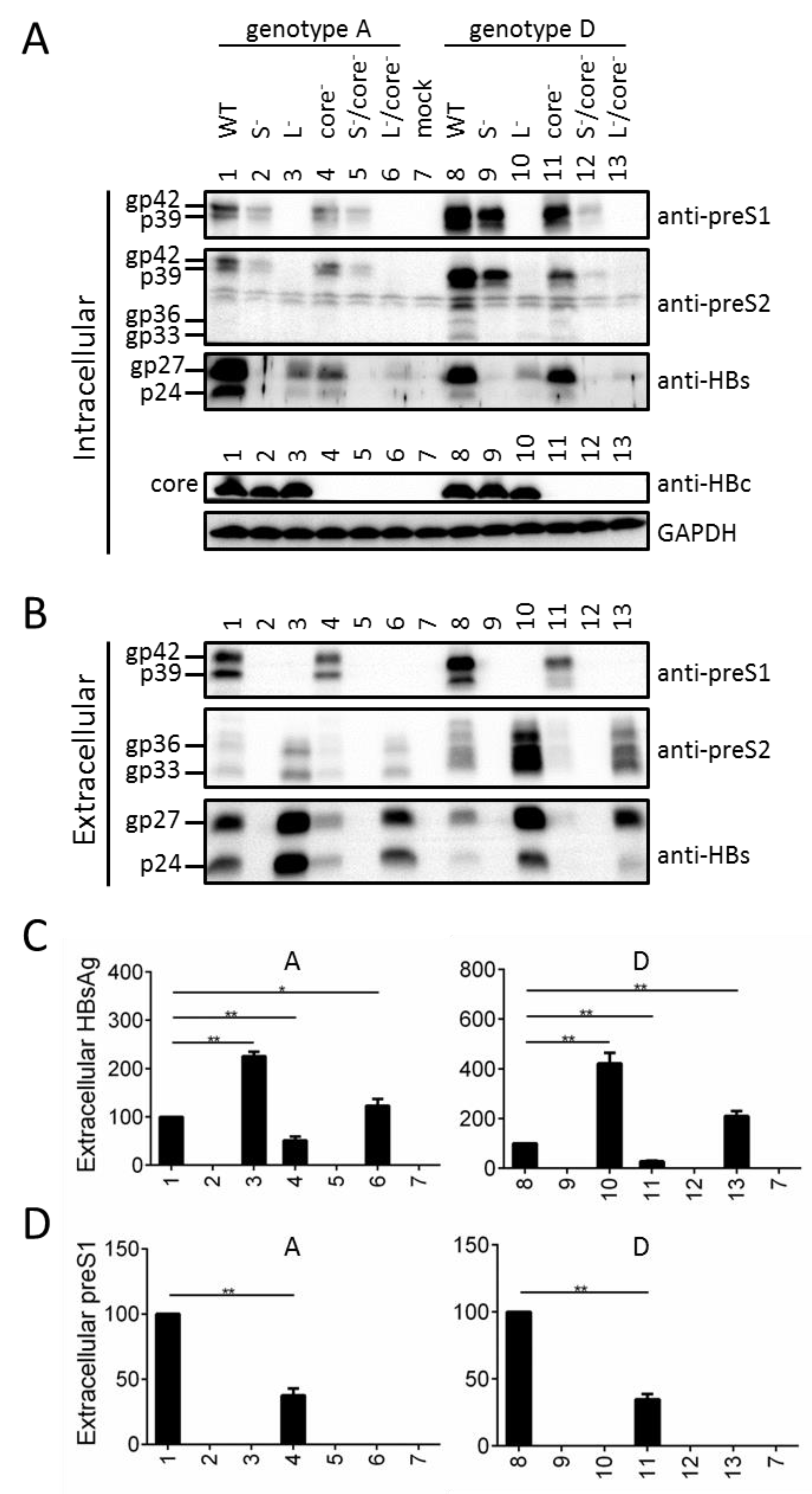
3.8. Preventing Core Protein Expression from the 1.1 mer Construct Reduced Intracellular and Extracellular Levels of Envelope Proteins, for Both Genotype A and Genotype D
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruss, V. Hepatitis B virus morphogenesis. World J. Gastroenterol. 2007, 13, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondot, M.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef] [PubMed]
- Bruss, V. A short linear sequence in the pre-S domain of the large hepatitis B virus envelope protein required for virion formation. J. Virol. 1997, 71, 9350–9357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schittl, B.; Bruss, V. Mutational profiling of the variability of individual amino acid positions in the hepatitis B virus matrix domain. Virology 2014, 458–459, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Standring, D.N.; Ou, J.; Rutter, W.J. Assembly of viral particles in xenopus oocytes: Pre-surface-antigens regulate secretion of the hepatitis B viral surface envelope particle. Proc. Natl. Acad. Sci. USA 1986, 83, 9338–9342. [Google Scholar] [CrossRef] [Green Version]
- Chisari, F.V.; Filippi, P.; McLachlan, A.; Milich, D.R.; Riggs, M.; Lee, S.; Palmiter, R.D.; Pinkert, C.A.; Brinster, R.L. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. J. Virol. 1986, 60, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Persing, D.H.; Varmus, H.E.; Ganem, D. Inhibition of secretion of hepatitis B surface antigen by a related presurface polypeptide. Science 1986, 234, 1388–1391. [Google Scholar] [CrossRef]
- Ou, J.H.; Rutter, W.J. Regulation of secretion of the hepatitis B virus major surface antigen by the preS-1 protein. J. Virol. 1987, 61, 782–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, T.; Li, J.; Sureau, C.; Ito, K.; Qin, Y.; Wands, J.; Tong, S. Drastic reduction in the production of subviral particles does not impair hepatitis B virus virion secretion. J. Virol. 2009, 83, 1152–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernholz, D.; Stemler, M.; Brunetto, M.; Bonino, F.; Will, H. Replicating and virion secreting hepatitis B mutant virus unable to produce preS2 protein. J. Hepatol. 1991, 13 (Suppl. 4), S102–S104. [Google Scholar] [CrossRef]
- Sureau, C.; Guerra, B.; Lee, H. The middle hepatitis B virus envelope protein is not necessary for infectivity of hepatitis delta virus. J. Virol. 1994, 68, 4063–4066. [Google Scholar] [CrossRef] [Green Version]
- Bruss, V.; Ganem, D. The role of envelope proteins in hepatitis B virus assembly. Proc. Natl. Acad. Sci. USA 1991, 88, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Fu, S.; Zhang, J.; Yuan, Q.; Li, J.; Xia, N.; Wen, Y.; Wang, Y.; Tong, S. Expression level of small envelope protein in addition to sequence divergence inside its major hydrophilic region contributes to more efficient surface antigen secretion by hepatitis B virus subgenotype D2 than subgenotype A2. Viruses 2020, 12, 967. [Google Scholar] [CrossRef]
- Zhang, F.; Tang, X.; Garcia, T.; Lok, A.S.; Wang, Y.; Jia, H.; Qin, Y.; Chen, C.; Wen, Y.; Li, J.; et al. Characterization of contrasting features between hepatitis B virus genotype A and genotype D in small envelope protein expression and surface antigen secretion. Virology 2017, 503, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Galibert, F.; Mandart, E.; Fitoussi, F.; Tiollais, P.; Charnay, P. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 1979, 281, 646–650. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Chen, S.; Yuan, Q.; Zhang, J.; Wu, J.; Jiang, Q.; Wang, Q.; Xia, N.; Zhang, J.; et al. Naturally occurring 5′preS1 deletions markedly enhance replication and infectivity of HBV genotype B. and genotype C. Gut 2020, 70, 575–584. [Google Scholar] [CrossRef]
- Sominskaya, I.; Paulij, W.; Jansons, J.; Sobotta, D.; Dreilina, D.; Sunnen, C.; Meisel, H.; Gerlich, W.H.; Pumpens, P. Fine-mapping of the B-cell epitope domain at the N-terminus of the preS2 region of the hepatitis B surface antigen. J. Immunol. Methods 2002, 260, 251–261. [Google Scholar] [CrossRef]
- Qin, Y.; Tang, X.; Garcia, T.; Hussain, M.; Zhang, J.; Lok, A.; Wands, J.; Li, J.; Tong, S. Hepatitis B virus genotype C isolates with wild-type core promoter sequence replicate less efficiently than genotype B isolates but possess higher virion secretion capacity. J. Virol. 2011, 85, 10167–10177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Gewaily, D.; Ahn, S.H.; Preskill, C.; Wang, Y.; Zong, L.; Zhang, J.; Han, K.; Wands, J.; Li, J.; et al. Sequence analysis and functional characterization of full-length hepatitis B virus genomes from Korean cirrhotic patients with or without liver cancer. Virus Res. 2017, 235, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zong, L.; Wang, Y.; Li, C.; Chen, C.; Wen, Y.; Li, J.; Tong, S. Core gene insertion in hepatitis B virus genotype G functions at both the encoded amino acid sequence and RNA structure levels to stimulate core protein expression. Virology 2019, 526, 203–213. [Google Scholar] [CrossRef]
- Kwei, K.; Tang, X.; Lok, A.S.; Sureau, C.; Garcia, T.; Li, J.; Wands, J.; Tong, S. Impaired virion secretion by hepatitis B virus immune escape mutants and its rescue by wild-type envelope proteins or a second-site mutation. J. Virol. 2013, 87, 2352–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponsel, D.; Bruss, V. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J. Virol. 2003, 77, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Qin, Y.; Zhang, J.; Jia, L.; Fu, S.; Wang, Y.; Li, J.; Tong, S. Tracing the evolutionary history of hepadnaviruses in terms of e antigen and middle envelope protein expression or processing. Virus Res. 2020, 276, 197825. [Google Scholar] [CrossRef]
- Chen, C.H.; Changchien, C.S.; Lee, C.M.; Hung, C.H.; Hu, T.H.; Wang, J.H.; Wang, J.C.; Lu, S.N. Combined mutations in pre-s/surface and core promoter/precore regions of hepatitis B virus increase the risk of hepatocellular carcinoma: A case-control study. J. Infect Dis. 2008, 198, 1634–1642. [Google Scholar] [CrossRef]
- Santantonio, T.; Jung, M.C.; Schneider, R.; Fernholz, D.; Milella, M.; Monno, L.; Pastore, G.; Pape, G.R.; Will, H. Hepatitis B virus genomes that cannot synthesize pre-S2 proteins occur frequently and as dominant virus populations in chronic carriers in Italy. Virology 1992, 188, 948. [Google Scholar] [CrossRef]
- Chen, B.F.; Liu, C.J.; Jow, G.M.; Chen, P.J.; Kao, J.H.; Chen, D.S. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology 2006, 130, 1153–1168. [Google Scholar] [CrossRef]
- Huy, T.T.; Ushijima, H.; Win, K.M.; Luengrojanakul, P.; Shrestha, P.K.; Zhong, Z.H.; Smirnov, A.V.; Taltavull, T.C.; Sata, T.; Abe, K. High prevalence of hepatitis B virus pre-s mutant in countries where it is endemic and its relationship with genotype and chronicity. J. Clin. Microbiol. 2003, 41, 5449–5455. [Google Scholar] [CrossRef] [Green Version]
- Raimondo, G.; Costantino, L.; Caccamo, G.; Pollicino, T.; Squadrito, G.; Cacciola, I.; Brancatelli, S. Non-sequencing molecular approaches to identify preS2-defective hepatitis B virus variants proved to be associated with severe liver diseases. J. Hepatol. 2004, 40, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs JA, G.; et al. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 2017, 22, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, N.; Locarnini, S. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatology 2008, 48, 88–98. [Google Scholar] [CrossRef]
- Ito, K.; Qin, Y.; Guarnieri, M.; Garcia, T.; Kwei, K.; Mizokami, M.; Zhang, J.; Li, J.; Wands, J.R.; Tong, S. Impairment of hepatitis B virus virion secretion by single-amino-acid substitutions in the small envelope protein and rescue by a novel glycosylation site. J. Virol. 2010, 84, 12850–12861. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N51 | nt ATG………………ATG……TCG…ATG…………CGCTGGATGTGTCTG………………………TAA aa M M S M R W M C L * |
| N65 | nt ATG………………ATG……TAG…ATG…………CGCTGGATGTGTCTG………………………TAA aa * |
| N65-tS | nt ATG………………ATG……TAG…GCG…………CGCTGGATGTGTCTG………………………TAA aa * A |
| N65-tS+ | nt ATG………………ATG……TAG…GCG…………CGCAGGATGGGTCTG………………………TAA aa * A R G |
| N65-nS | nt ATG………………ATG……TAG…GCG…………CGCTGGGCGTGTCTG………………………TAA aa * A A |
| N67 | nt ATG………………ATG……TCG…GCG…………CGCTGGATGTGTCTG………………………TAA aa A |
| N67+ | nt ATG………………ATG……TCG…GCG…………CGCAGGATGGGTCTG………………………TAA aa A R G |
| N67-ns | nt ATG………………ATG……TCG…GCG…………CGCTGGGCGTGTCTG………………………TAA aa A A |
| N67a | nt ATG………………ATG……TCG…ATA…………CGCTGGATGTGTCTG………………………TAA aa I |
| N67a+1 | nt ATG………………ATG……TCG…ATA…………CGCGGGATGGGTCTG………………………TAA aa I G G |
| N67a+2 | nt ATG………………ATG……TCG…ATA…………CGCAGGATGGGTCTG………………………TAA aa I R G |
| N67a- | nt ATG………………ATG……TCG…ATA…………CGTTTTATGTGTCTT………………………TAA aa I F |
| N67a-nS | nt ATG………………ATG……TCG…ATA…………CGCTGGGCGTGTCTG………………………TAA aa I A |
| N67b | nt ATG………………ATG……TCG…AAG…………CGCTGGATGTGTCTG………………………TAA aa K |
| N67b+1 | nt ATG………………ATG……TCG…AAG…………CGCGGGATGGGTCTG………………………TAA aa K G G |
| N67b+2 | nt ATG………………ATG……TCG…AAG…………CGCAGGATGGGTCTG………………………TAA aa K R G |
| N67b- | nt ATG………………ATG……TCG…AAG…………CGTTTTATGTGTCTT………………………TAA aa K F |
| N67b-nS | nt ATG………………ATG……TCG…AAG…………CGCTGGGCGTGTCTG………………………TAA aa K A |
| N67c | nt ATG………………ATG……TCG…TTG…………CGCTGGATGTGTCTG………………………TAA aa L |
| Mutation | Genotype (Clone) | Change (nt) | Change (aa) | Change (aa, Overlapping Gene) |
|---|---|---|---|---|
| core | A (geno5.4) | C2044A | Core: C48* | N/A |
| core | D (geno1.2) | T2044A | Core: C48* | N/A |
| L | A (geno5.4) | G3008A | L: W52* | Pol: silent |
| L | D (geno1.2) | A2959T | L: K38* | Pol: Q218L |
| S | A (geno5.4) | T156C | S: M1T | Pol: silent |
| S | D (geno1.2) | T156C | S: M1T | Pol: silent |
| S | D (#) | T156C | S: M1T | Pol: silent |
| AAAA (preS2) | D (#) | ## | preS2: Q13A/D14A/P15A/R16A | Pol: A301G/Q304G |
| del13–16 (preS2) | D (#) | del26CAAGATCCCAGA37 | preS2: del13QDPR16 | Pol: del301ARSQ304/S305G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Wang, Y.; Fu, S.; Yuan, Q.; Wang, Q.; Xia, N.; Wen, Y.; Li, J.; Tong, S. Role of Small Envelope Protein in Sustaining the Intracellular and Extracellular Levels of Hepatitis B Virus Large and Middle Envelope Proteins. Viruses 2021, 13, 613. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040613
Zhang J, Wang Y, Fu S, Yuan Q, Wang Q, Xia N, Wen Y, Li J, Tong S. Role of Small Envelope Protein in Sustaining the Intracellular and Extracellular Levels of Hepatitis B Virus Large and Middle Envelope Proteins. Viruses. 2021; 13(4):613. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040613
Chicago/Turabian StyleZhang, Jing, Yongxiang Wang, Shuwen Fu, Quan Yuan, Qianru Wang, Ningshao Xia, Yumei Wen, Jisu Li, and Shuping Tong. 2021. "Role of Small Envelope Protein in Sustaining the Intracellular and Extracellular Levels of Hepatitis B Virus Large and Middle Envelope Proteins" Viruses 13, no. 4: 613. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040613