1. Introduction
Hepatitis delta virus (HDV) is a satellite virus that requires hepadnavirus envelope proteins for its transmission. Approximately 5% of Hepatitis B Virus (HBV) carriers have been exposed to HDV, with a total of 15–20 million patients worldwide, although recent studies have reported higher prevalence numbers [
1,
2,
3]. HDV infection is associated with the most severe form of viral hepatitis, with a twofold higher risk of developing cirrhosis, a threefold higher risk of developing hepatocellular carcinoma (HCC), and twofold increased mortality in comparison with HBV monoinfection [
4,
5,
6]. Despite recent advances in the management of this condition, it still represents a significant medical burden [
6,
7,
8]. Conventional treatment for chronic HDV infection consists of long-term administration of standard or pegylated interferon alpha (peg-IFN-α), which leads to a sustained virological response (SVR) in only 20–30% of patients treated and is frequently associated with serious adverse reactions. Several new agents are now under clinical investigation: bulevirtide, lonafarnib and nucleic acid polymers [
9,
10]. These drugs target essential steps of the HDV viral cycle - like viral entry, the isoprenylation of the large viral antigen, and viral encapsidation. Importantly, bulevirtide was approved in Europe in July 2020 [
11].
The mechanisms associated with the severity of the disease remains unknown, although it is thought to be associated with the host immune response since a significant inflammatory infiltrate composed of macrophages and lymphocytes is observed in the liver of HDV patients [
12,
13]. However, HDV has been also considered to be directly cytopathic, particularly during the acute stage of infection, and it has been related to the expression of HDV antigens (HDAgs) [
14,
15,
16]. The cytotoxic properties of HDAgs are largely unsettled due to the different results obtained in vitro, which depend on the cellular systems employed [
15,
16,
17]
HDV is a satellite RNA virus that requires the surface antigens of HBV (HBsAg) for viral assembly and transmission [
10]. Specific interaction between HBsAg and the human Na
+/taurocholate co-transporting polypeptide (hNTCP) determines the hepatotropism and species-specificity of both viruses [
18,
19]. Although NTCP is also expressed in mouse livers, mice are resistant to HBV and HDV infections due to small differences between the human and mouse NTCP protein sequences [
18].
The HDV genome is a circular negative-sense RNA molecule of approximately 1700 nucleotides that appears as a double-stranded rod-like structure. It contains a single open reading frame (ORF) that encodes two HDAgs—a 24 kDa small HDV antigen (S-HDAg), and a 27 kDa large HDV antigen (L-HDAg) of 195 and 214 amino acids, respectively [
10]. Despite sharing most of their amino acid sequence, they differ significantly in their functions: it has been described that S-HDAg is essential for HDV replication, whilst L-HDAg blocks HDV replication and is essential for viral assembly [
17,
20,
21]. The production of L-HDAg requires RNA editing at adenosine 1012 (amber/W site) in the antigenomic RNA sequence by host adenosine deaminase that act on RNA-1 (ADAR-1) [
22].
All these studies have been performed in cell culture due to the lack of an in vivo model in which the HDV genome can be easily manipulated and tested [
23]. Recently, we developed a mouse model of HDV replication, based on the adeno-associated viral vector (AAV)-mediated delivery of HBV and HDV replication-competent genomes to the liver. This model mimics most of the features of HDV infection in humans, including the induction of liver inflammation and liver injury in association with the expression of genes involved in the development of HCC, cirrhosis, fibrosis, and cell death [
24]. Using this model, we described that mitochondrial antiviral signalling protein (MAVS) was responsible for the strong type I interferon (IFN) response triggered by HDV replication, observed in cell culture and animal models [
23,
24,
25,
26,
27]. This result was corroborated in human hepatic cells identifying melanoma differentiation-associated protein 5 (MDA5) as the main cellular pattern recognition receptor (PRR) involved in HDV detection that signals through MAVS and induces IFN-β gene transcription [
28]. Furthermore, the involvement of tumor necrosis factor-alpha (TNF-α) in the HDV-induced pathology was demonstrated using this animal model and it was confirmed by the amelioration of liver damage in AAV-HBV/HDV-injected animals treated with Etanercept, a drug that blocks TNF-α receptor interaction [
29].
One of the beauties of this model is that it allows for genetic modification of HDV viral genomes that can be then tested in cell culture and more importantly in the liver of mice to interrogate the role of viral components in the virus life cycle and in the induction of liver pathology. Furthermore, the establishment of HDV replication in genetically manipulated mice allows us to analyze the involvement of different host factors.
The aim of this work was to determine the role of HDV antigens in HDV-mediated liver injury using AAV-HDV carrying different mutations.
2. Materials and Methods
2.1. Plasmids and AAV Vectors
The AAV-HDV wild-type (WT) plasmid was generated as previously described by cloning the HDV 1.2× (genotype 1) genome sequence obtained from the plasmid pDL456 (kindly provided by J. M. Taylor) under the control of the hepato-specific promoter—enhancer of albumin/alpha-1-antitrypsin promoter (EAlb/AAT)—into the AAV-MCS plasmid [
24]. Different HDV mutants were created by introducing point mutations. All the plasmids were generated with the In-Fusion
® HD Cloning Kit (Takara Bio USA, #638910, Mountain View, CA, USA) following the manufacturer’s instructions. To that end, the HDV-ΔS-HDAg, HDV-ΔL-HDAg, and HDV-ΔHDAg mutants were produced to block the expression of either S-HDAg, L-HDAg, or both, respectively. The first mutant contains a single nucleotide change (A/G) at nucleotide 1015 in the editable amber stop codon (TAG), which produces a tryptophan codon (TGG) that cannot be edited, always leading to the production of L-HDAg. The second mutant (HDV-ΔL-HDAg) contains a point mutation (G/A) at nucleotide 1016 that changes the amber/W site (TAG) into another stop codon (TAA), which cannot be converted into a tryptophan codon by ADAR-1 [
21]. Therefore, from this sequence, only S-HDAg will be produced. For the construction of the HDV-ΔHDAg mutant, the start codons found in the sequence (located between nucleotides 1088–1090 and 1364–1366) were replaced by stop codons (TAA). Finally, AAV-HBV plasmid was constructed by insertion of 1.3× copies of the HBV genome (genotype D, serotype ayw) obtained from the pSP65 plasmid (kindly provided by Dr. Francis Chisari) into pAAV-MCS [
24].
The recombinant AAV genomes were encapsidated in the mouse liver tropic AAV serotype 8 capsid. Briefly, each AAV vector plasmid and the helper/packaging plasmid pDP8.ape (Plasmid factory, Bielefeld, Germany) were co-transfected into HEK-293T cells. The cells and supernatants were harvested 72 h after transfection, and the virus was released from the cells by three rounds of freeze-thawing. Crude lysate from all batches was then treated with DNAse and RNAse (0.1 mg per p150 culture dish) for 1 h at 37 °C and then kept at −80 °C until purification. Purification of crude lysate was performed by ultracentrifugation in optiprep density gradient medium-iodixanol (Sigma-Aldrich, St Louis, MO, USA). Thereafter, iodioxanol was removed, and the batches were concentrated by passage through Amicon Ultra-15 tubes (Ultracel-100K; Merck Millipore, Cork Ireland). For virus titration, viral DNA was isolated using the High Pure Viral Nucleic Acid kit (Roche Applied Science, Mannheim, Germany). Viral titers in terms of viral genome copies per millilitre (vg/mL) were determined by Real Time-qPCR (#1855196, BioRad Hercules, CA, USA).
2.2. Animals and Treatment
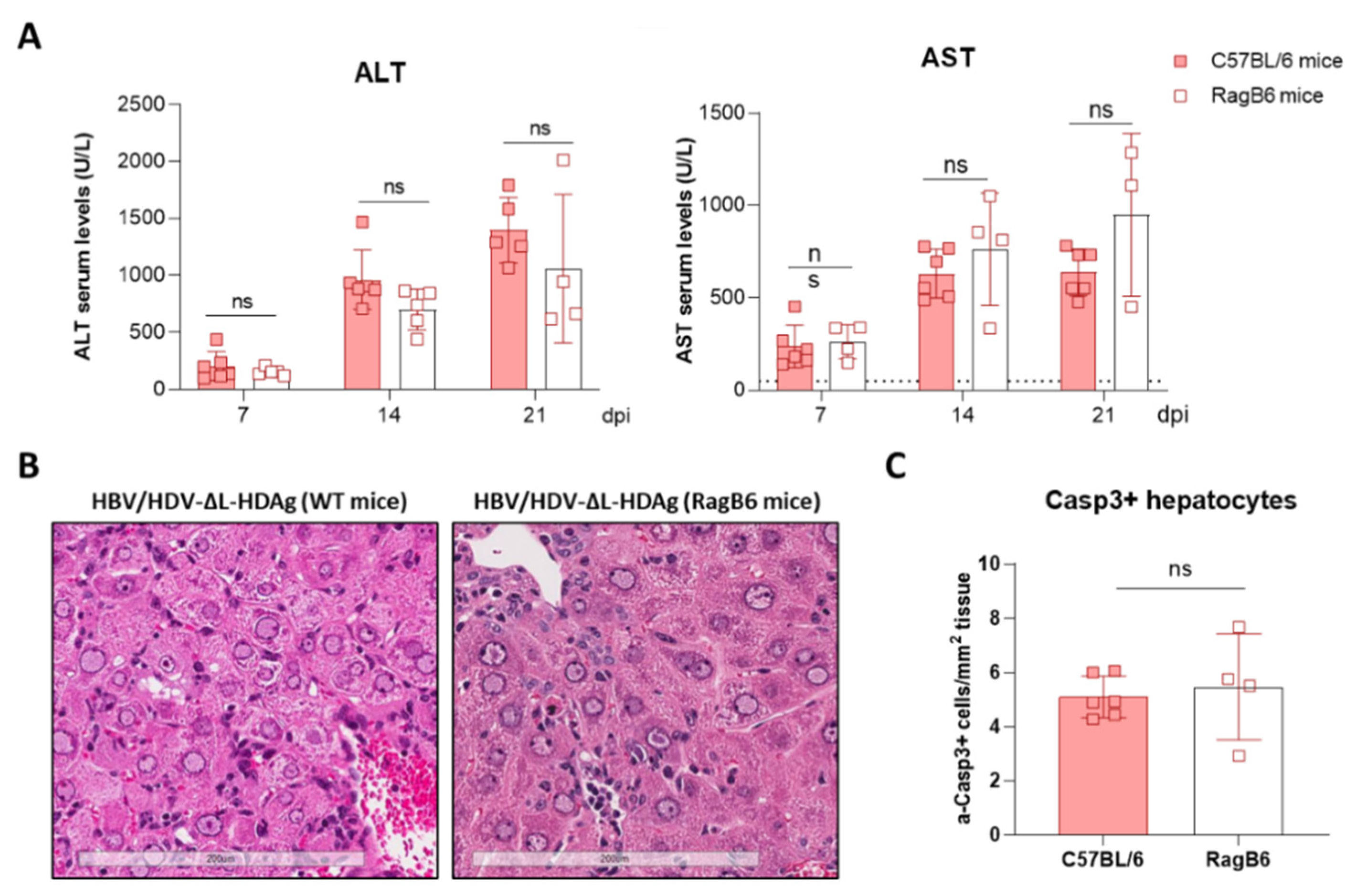
C57BL/6 mice were purchased from Harlan Laboratories (Barcelona, Spain). Recombination activating gene 1 (RAG-1) deficient mice on a C57BL/6 genetic background (RagB6) were bred and maintained at the animal facility of the University of Navarra. Six- to eight-week-old male mice were used in all experiments. Mice were kept under controlled temperature, light, and pathogen-free conditions. Mice were injected intravenously (i.v.) with AAV vectors (5 × 1010 vg of each vector per mouse) diluted in saline solution in a volume of 100 µL. For all procedures, animals were anesthetized by intraperitoneal (i.p.) injection of a mixture of xylazine (Rompun 2%, Bayer) and ketamine (Imalgene 50, Merial) 1:9 v/v or by isoflurane inhalation. Blood collection was performed by submandibular bleeding, and serum samples were obtained after centrifugation of total blood. Animals were euthanized by cervical dislocation after being anesthetized. The experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra (R-132-19GN).
2.3. Cell Lines
Two human hepatoma cell lines, HepG2 (ATCC® HB-8065™) and Huh-7 (ATCC® PTA-4583), were transfected with HDV WT-encoding plasmids, while the characterization of HDV-ΔL-HDAg, HDV-ΔS-HDAg, and HDV-ΔHDAg mutants was only performed in Huh-7 cells. Moreover, Huh-7-hNTCP cells, kindly provided by Dr. Urtzi Garaigorta, were employed for infectivity studies. HepG2 and Huh-7 cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% of L-glutamine, 1% of glucose, 100 U/mL of penicillin-streptomycin, and non-essential amino acids, and incubated at 37 °C with 5% CO2 in a humidified atmosphere. In the case of Huh-7-hNTCP cells, the culture medium was supplemented with 2.5 µg/mL blasticidine to ensure the selection of hNTCP-expressing cells. HEK293T cells (ATCC® PTA-4583) were used for AAV production.
2.4. DNA Transfection
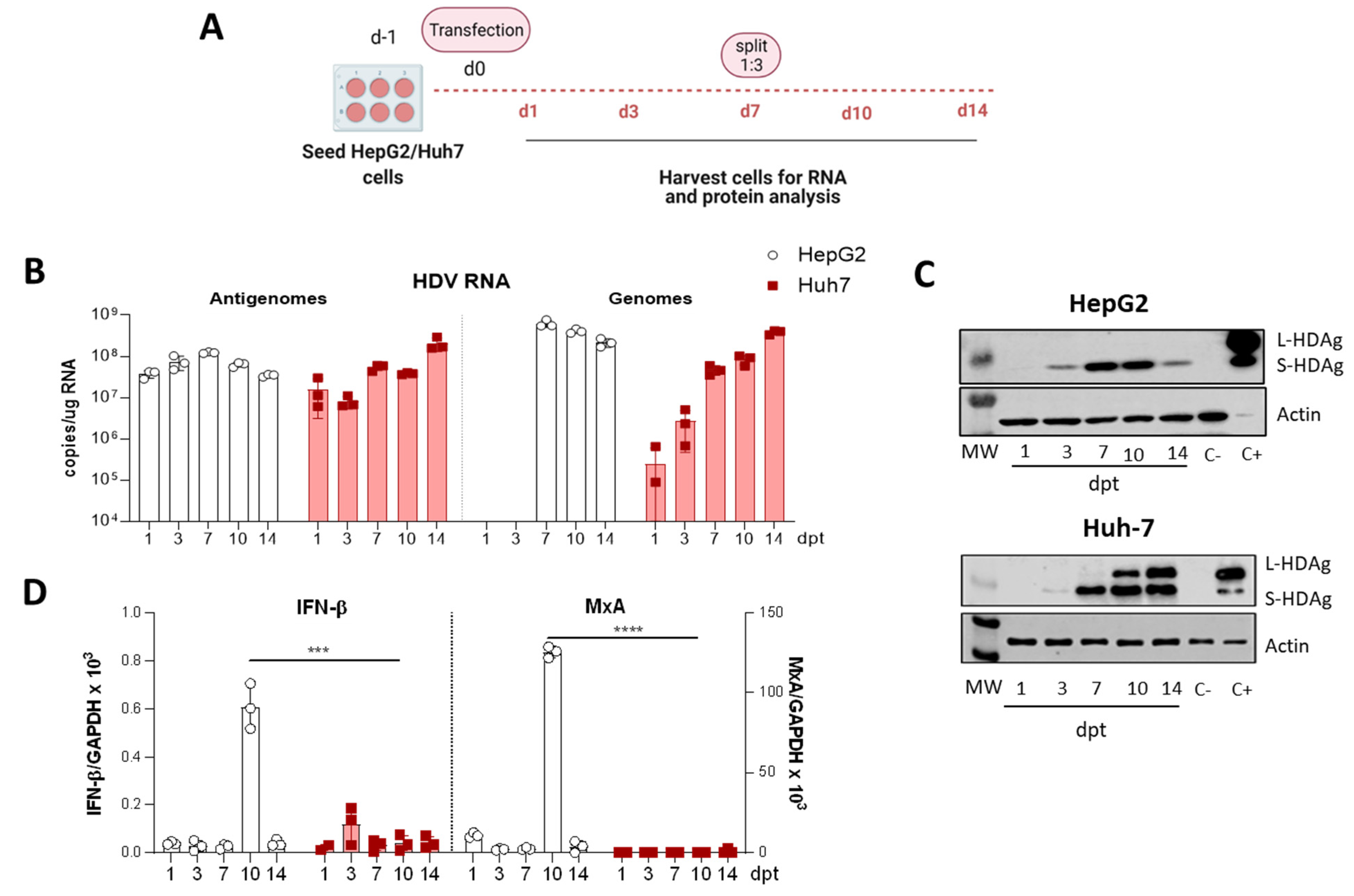
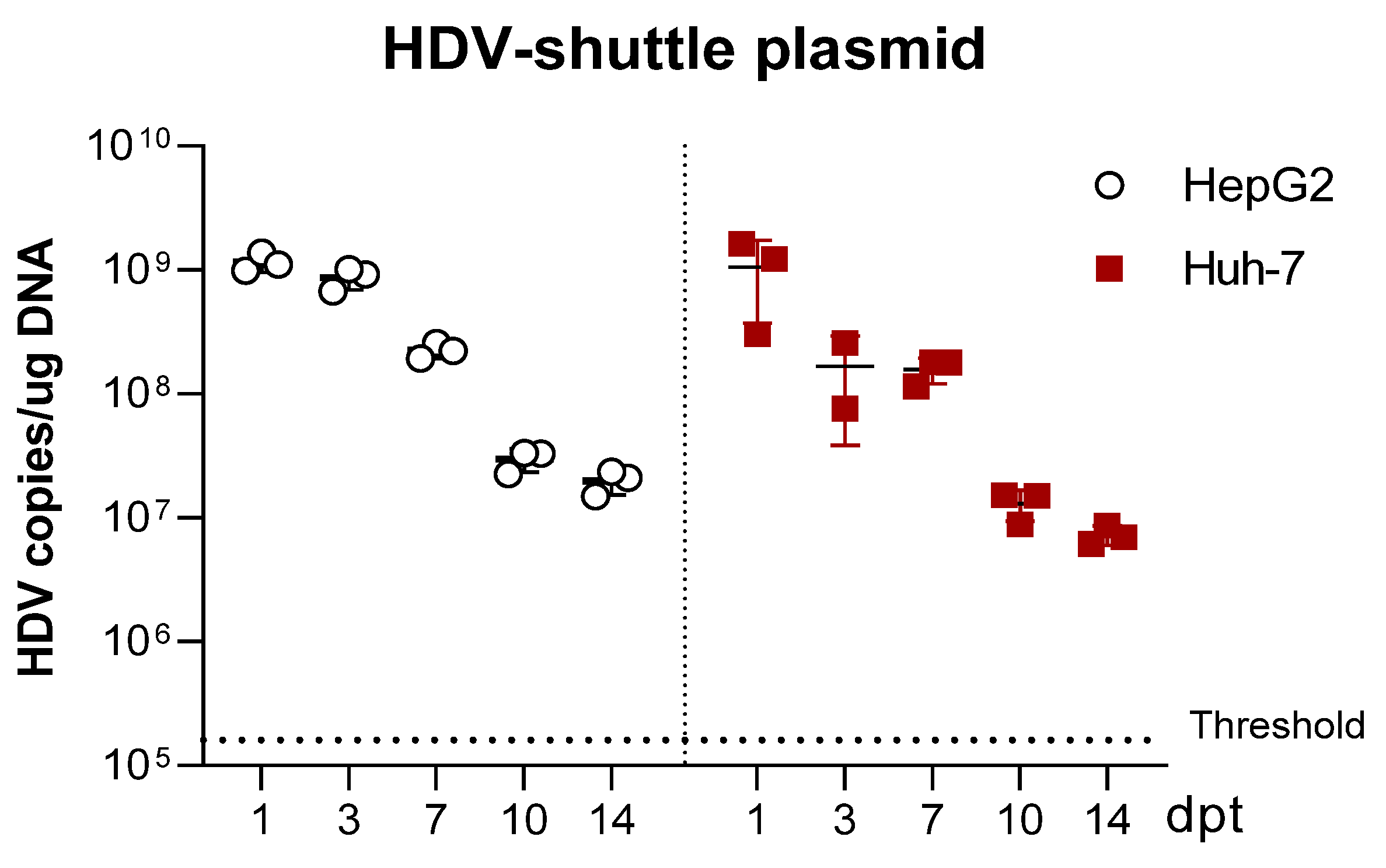
For the comparative study of HDV transfection in HepG2 and Huh-7 cells, both cell lines were seeded and transfected in parallel. Briefly, 3.5 × 105 HepG2 and Huh-7 cells were seeded per well in 6-well plates and maintained in DMEM 10% FBS. After 24h, the medium was substituted by Opti-mem, and cells were transfected with 2.5 µg DNA/well using Lipofectamine 3000, following the manufacturer’s instructions. 4–6 h later, cells were supplemented with 1.5 mL of DMEM 10% FBS, and the day after, the medium containing the transfection reagents was replaced by fresh DMEM 10% FBS. Cells were then collected at different time points until 14 days post-transfection (dpt). At 7-dpt, cells were trypsinized, split 1:3 and reseeded in new wells.
2.5. Cell Fractionation, Protein Extraction and Quantification
Harvested cells were resuspended in the RIPA lysis buffer supplemented with 1mM PMSF (phenylmethylsulfonyl fluoride), 1% v/v of a pre-formed protease inhibitor cocktail (#P8340, Sigma-Aldrich: 4-(2-Aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF) at 104 mM, Aprotinin at 80 μM, Bestatin at 4 mM, E-64 at 1.4 mM, Leupeptin at 2 mM and Pepstatin A at 1.5 mM), and 0.1 mM of sodium orthovanadate, and incubated on ice for 30 min. Cell extracts were then spun for 20 min at 13,000 rpm and 4 °C, and supernatants were collected for western blot analysis. Protein concentration was determined by BCA assay (#23227, Thermo Fisher Scientific, Waltham, MA, USA).
To determine the intracellular location of the proteins, cells were harvested with Trypsin-EDTA and centrifuged for 5 min at 500 g and 4 °C. Then, proteins of each cellular compartment were separated with the Subcellular Protein Fractionation Kit (#78840, Thermo Fisher Scientific), following the manufacturer’s instructions.
2.6. Western Blot
Equal amounts of protein were loaded in a SDS-polyacrylamide gel, separated by electrophoresis, and transferred onto a nitrocellulose membrane by electroelution. After a 45 min blockade with TBS-Tween™ 20 with 5% non-fat dry milk at room temperature (RT), the membranes were incubated overnight at 4 °C with the serum from patient CUN-28336 at dilution 1:2500. Patient serum was provided by the Biobank of the University of Navarra and processed following standard operating procedures approved by the Ethical and Scientific Committee (2019.217 CEI-CUN). After an extensive wash with TBS-Tween™ 20, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at RT for 1h. The substrate of the enzyme was provided by the reagents of SuperSignal™ West Femto (#34095, Thermo Fisher Scientific), and the signal was detected by the ODYSSEY CLx near-infrared fluorescence imaging system.
2.7. RNA Extraction and RT-qPCR
Total RNA from liver samples was isolated using TRI Reagent
® (#T9424, Sigma-Aldrich) according to the manufacturer’s instructions. Total RNA was pre-treated with DNAse I (#AM-1907, TURBO DNA-free™ Kit, Applied Biosystems, Foster City, CA, USA) and reverse-transcribed into complementary DNA (cDNA) using M-MLV reverse-transcriptase (Invitrogen, #28025013). Real-time quantitative polymerase chain reactions (RT-qPCR) were performed using iQ SYBR Green Supermix (#170-8884, BioRad) in a CFX96 Real-Time Detection System (#1855196, BioRad) and primers as specified in
Appendix A. HDV strand-specificity was analyzed as described elsewhere [
24]. GAPDH was used as a control housekeeping gene.
2.8. Immunofluorescence (IF)
Cells were fixed in PBS 4% paraformaldehyde (PFA) for 20 min at RT and permeabilized with PBS 0.1% Triton X-100 for 15 min at RT prior incubation with PBS-0.1% tween-20 5% BSA (blocking buffer) for 30 min at 37 °C. After washing, cells were incubated for 30 min at 37 °C with the serum from patient CUN-28336 at dilution 1:2500. Then, cells were incubated with the secondary antibody (goat anti-human IgG conjugated to Alexa Fluor 488, Applied Biosystems-Life Technologies, Waltham, MA, USA) at dilution 1:3000 for 30 min at 37 °C, protected from light. Finally, samples were covered with a mounting medium containing DAPI (1.5 µg/mL, Vector Laboratories), and the fluorescent samples were visualized with a confocal microscope (Zeiss LSM 880, Carl Zeiss, Oberkochen, Germany).
2.9. Histology and Immunohistochemistry (IHC)
Hematoxylin & Eosin (H&E): Liver sections were fixed with 4% paraformaldehyde (PFA), embedded in paraffin, sectioned (3 μm), and stained with hematoxylin and eosin. Sections were mounted and analysed by light microscopy for histologic evaluation.
Immunohistochemistry (IHC): the first steps were the same as for the H&E staining. Then, a step of antigen retrieval was performed that consisted of incubation for 30 min at 95 °C in 0.01 M Tris-1 mM EDTA pH 9. Subsequently, primary antibodies were incubated overnight at 4 °C. After rinsing in TBS-T, the sections were incubated with the corresponding secondary antibodies for 30 min at RT. Peroxidase activity was revealed using DAB+ and sections were lightly counterstained with Harris hematoxylin. Finally, slides were dehydrated in graded series of ethanol, cleared in xylene and mounted with Eukitt (Labolan, #28500, Navarra, Spain). Image acquisition was performed on an Aperio CS2 slide scanner using ScanScope Software (Leica Biosystems, Vista, CA, USA). The image analysis was performed using a plugin developed for Fiji, ImageJ (NIH, Bethesda, MD, USA). The antibodies employed are summarized in
Table 1.
2.10. Infectivity Studies
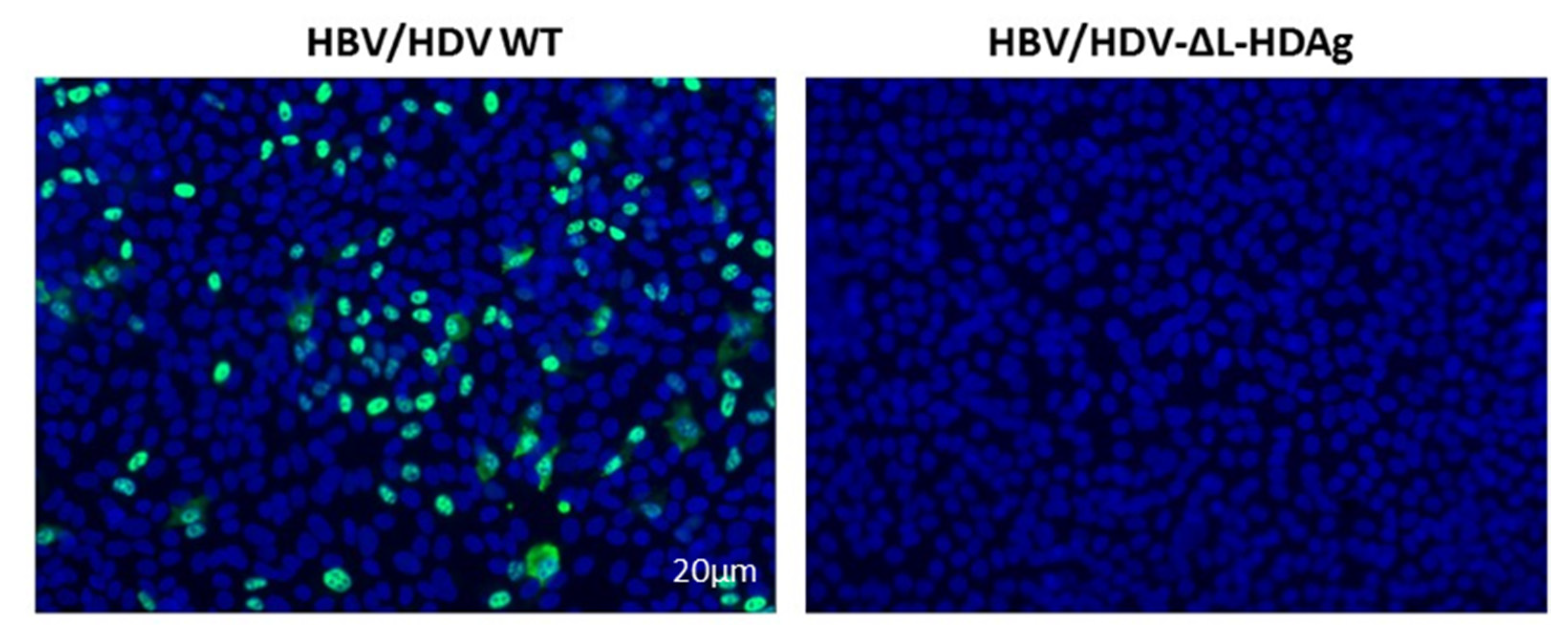
Huh-7-hNTCP cells were seeded in 12-well plates with coverslips and maintained in DMEM 10% FBS 2% DMSO. The day of infection, culture medium was replaced by 1 mL of supernatant collected from Huh-7 co-transfected with HBV/HDV WT or HBV/HDV-mutants at 7 and 14 dpt. The day after infection, the medium was replaced by fresh DMEM 2% FBS 2% DMSO, and at 7 dpi, cells were fixed in 4% PFA, and the presence of HDAg-positive cells was examined by performing IF, as indicated above.
2.11. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8.0 software. The data are presented as individual values ± standard deviation. Statistical significance was determined using an unpaired t-test for single comparisons and two-way ANOVA followed by Bonferroni’s multiple comparison tests to find differences between groups. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
4. Discussion
HDV infection is responsible for the most severe form of viral hepatitis in humans, leading to cirrhosis in 80% of cases within 10 years, and with a significant proportion of patients dying of hepatic decompensation [
1,
3]. The mechanism involved in the severity of the disease remains obscure, largely because of the lack of appropriated small animal models in which in vivo molecular analysis can be performed. Relevant animal models of HDV infection like chimpanzees or woodchucks developed liver damage that can even be fatal, but the performance of mechanistic studies in these animal models is cumbersome [
30,
31]. Recently, we have developed a HDV mouse model that overcomes species-related limitations based on the transfer of the HDV replication-competent genome to mouse hepatocytes via an AAV vector [
24]. For the first time, HDV-mediated liver injury was observed in mice, in association with a strong inflammatory infiltrate and hepatocyte apoptosis. Furthermore, we found that TNF-α was partially responsible for the observed liver damage. The beauty of this model is that animals with different genetic backgrounds can be used, and that mutations affecting specific viral components can be easily introduced in the HDV genome and tested in vivo [
24,
29].
Pioneering work performed in the ’80–’90s by several prominent virologists demonstrated the role of HDAgs in the viral cycle and the induction of cell damage [
14,
15,
20,
21,
32,
33,
34]. Studies performed using different cellular systems and transfection of plasmids carrying replication competent HDV genomes or expressing the HDAgs demonstrated the differential roles of the small and the large antigens on HDV biology. They showed that while S-HDAg is necessary for HDV genome replication, L-HDAg has the capacity to inhibit it [
32,
33,
34]. Furthermore, they also showed that the expression of S-HDAg might exert a cytotoxic effect and that L-HDAg interferes with multiple cellular signalling pathways and alters cellular homeostasis by inducing oxidative stress or increasing susceptibility to the effect of inflammatory cytokines [
34,
35,
36,
37,
38]. Here, using the AAV vector as an HDV delivery platform, we have tested in vivo the role of HDAg in the viral life cycle and in the induction of liver damage in mice.
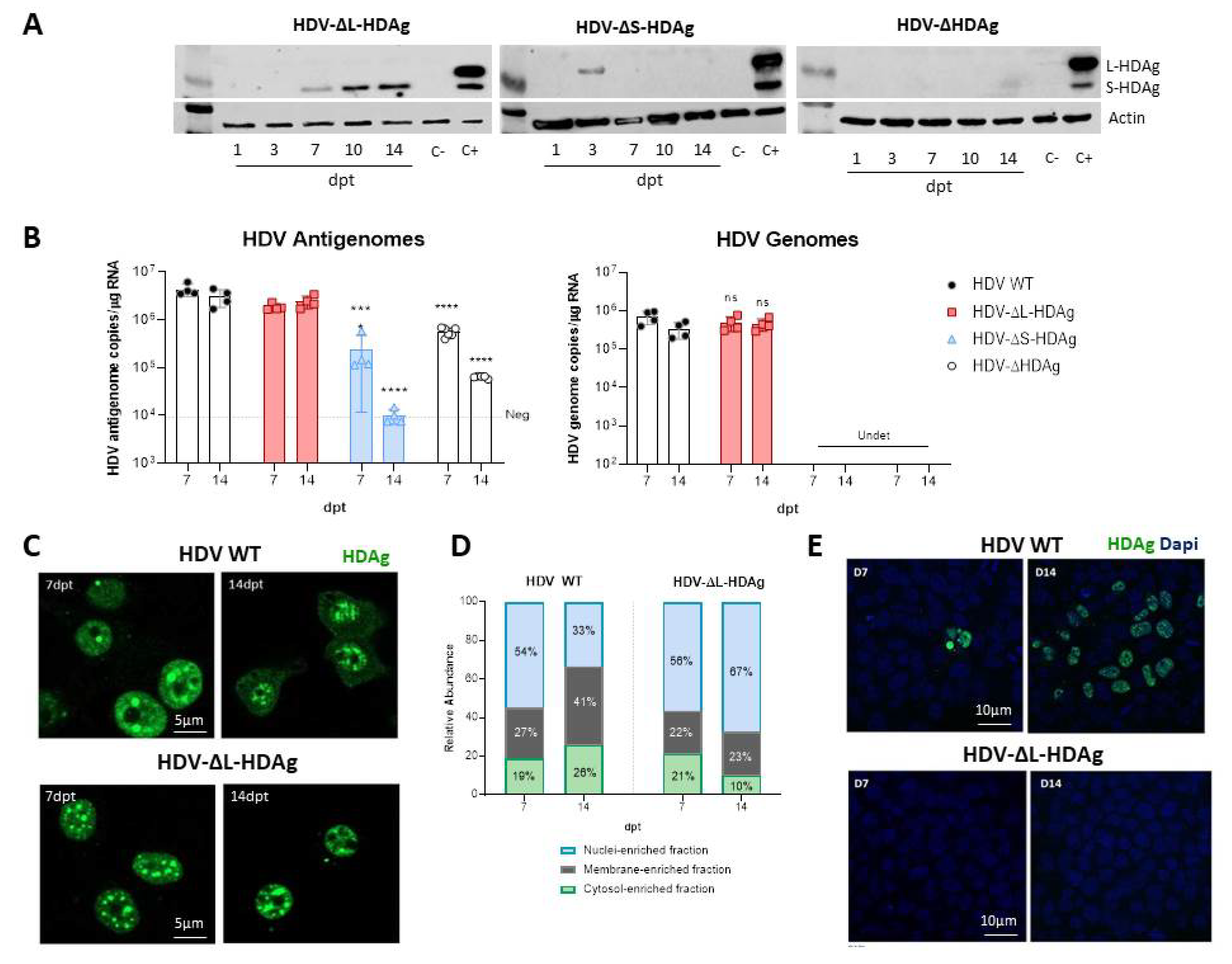
We have generated three different mutants with altered expression of HDAgs: one lacking HDAg expression, the second expressing only L-HDAg, and the third expressing only S-HDAg. The initial characterization of the mutants was performed in vitro by plasmid transfection into two hepatic cell lines of human origin. Firstly, HepG2 and Huh-7 cells were transfected with plasmids carrying the HDV WT sequence. We observed that in Huh-7 cells, both viral replication and HDAgs expression were sustained, despite cell passages and loss of the shuttle plasmid; however, in HepG2 cells, HDV replication and antigen expression decreased after splitting the cells. Furthermore, while both HDV antigens were expressed in Huh-7 cells, only S-HDAg was detected in HepG2. The absence of L-HDAg in HepG2 cells cannot be explained by the lack of ADAR-1, since both cell lines expressed similar levels of the editing protein (data not shown). One of the main differences we observed between the two cell lines is the activation of the cellular innate immune response to HDV replication. In HepG2 cells, we observed a strong activation of type I IFN response that was almost absent in Huh-7 cells. Interestingly, expression of IFN-β and MxA was detected only at day 10 after transfection, 3 days after splitting the cells. A potential explanation for this sharp peak is that at day 10, although undetectable by western blot, L-HDAg is present and HDV ribonucleoproteins (HDV RNPs) translocate from the nucleus to the cytoplasm where the viral RNA is sensed by MDA5 and initiates the activation of the innate immune response. Alternatively, the increase of the cellular division rate due to cell splitting might expose the RNPs to cytosolic RNA sensors.
Recent work by Zhang et al. has similarly shown that HDV-induced IFN response suppresses cell division mediated-HDV spread in HepaRG cells [
39], and this could explain why HDV replication is sustained in Huh-7 but not in HepG2 cells in our experimental setting.
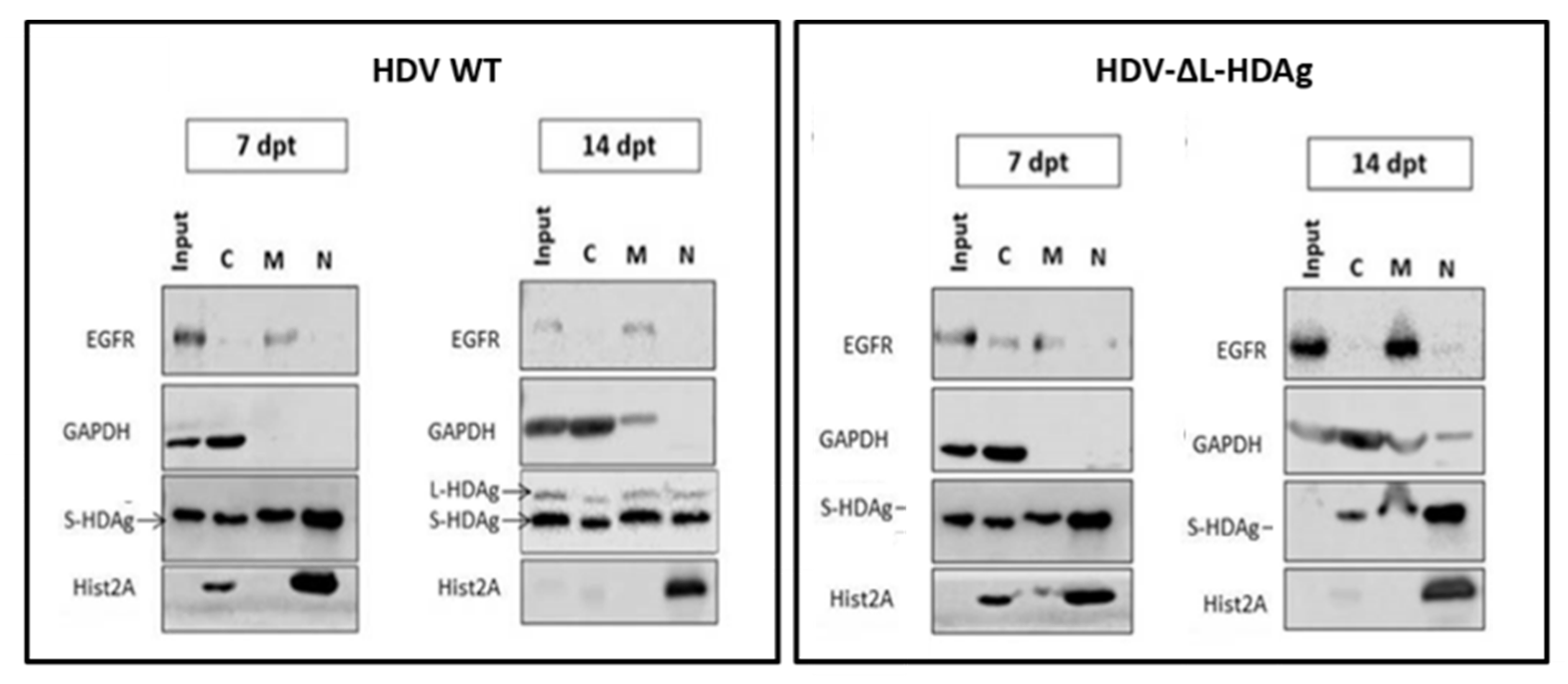
Both in vitro and in vivo studies showed that the HDV genome itself is unable to initiate replication and requires the expression of viral antigens, indicating that the interaction between the cellular polymerase and the viral genome cannot be replaced by a host protein. More interestingly, S-HDAg cannot be substituted by the large antigen despite both sequences being identical except for the 19 extra amino acids in the carboxy-terminal region of L-HDAg. On the other hand, we also confirmed in vitro that L-HDAg is essential for the export of the HDV RNP from the nucleus to the cytoplasm and for the formation of HDV infective particles, both in vitro and in vivo [
34,
40], and that these functions cannot be exerted by S-HDAg.
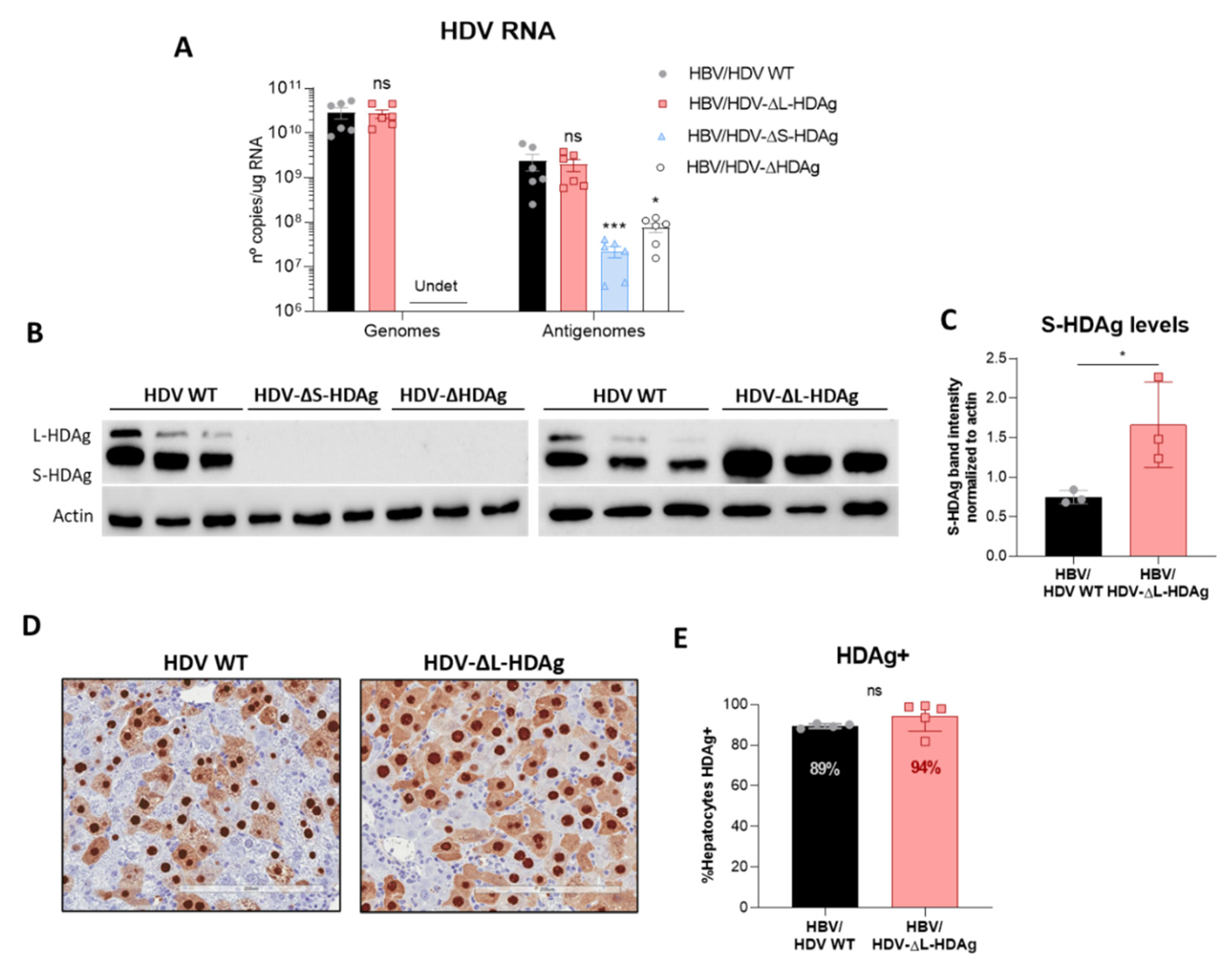
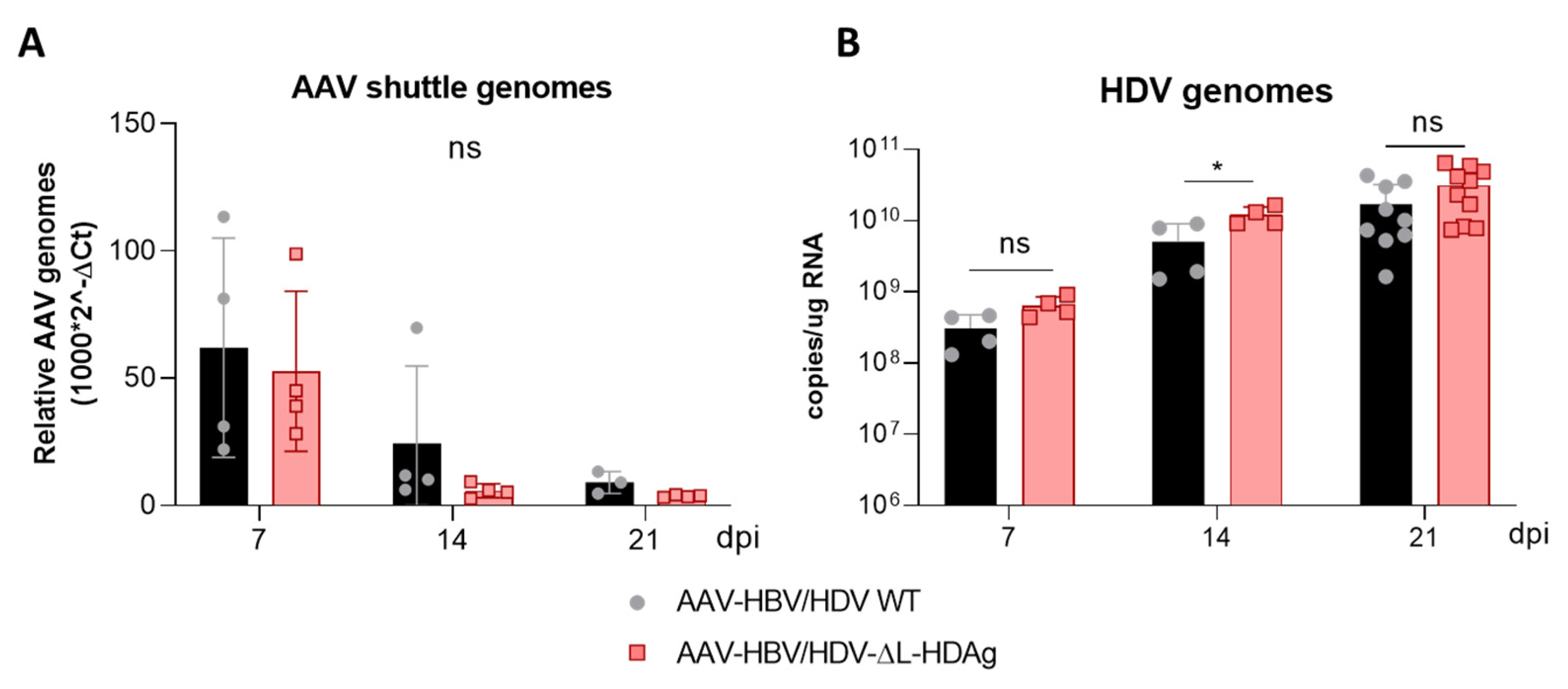
Interestingly, S-HDAg expression levels were significantly higher in the mutant lacking L-HDAg expression, and while viral replication was similar at 21 dpi, it was significantly higher at 14 dpi (
Figure A3), pointing towards the regulatory role of this isoform in the inhibition of HDV replication as previously reported in vitro [
21,
34,
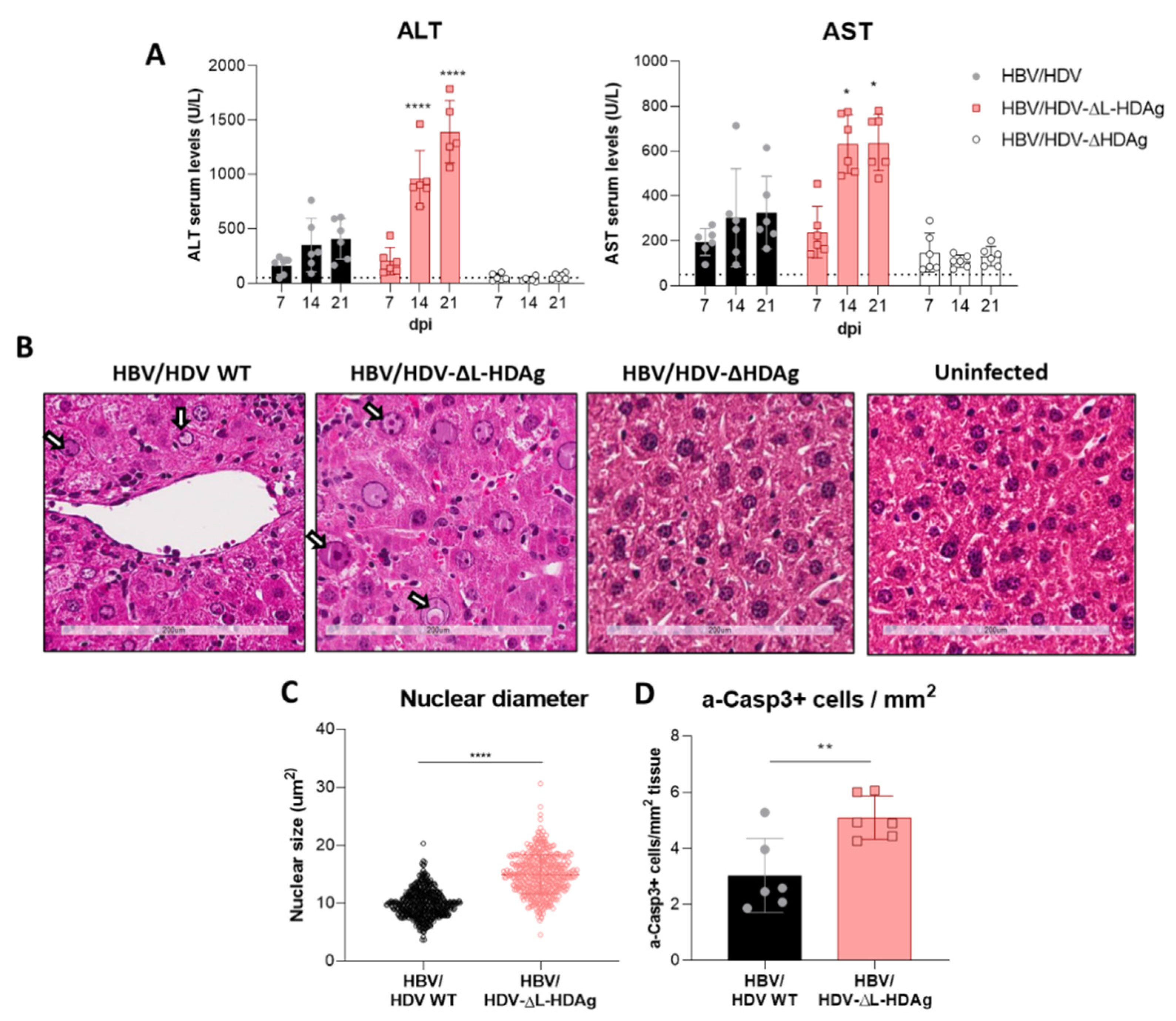
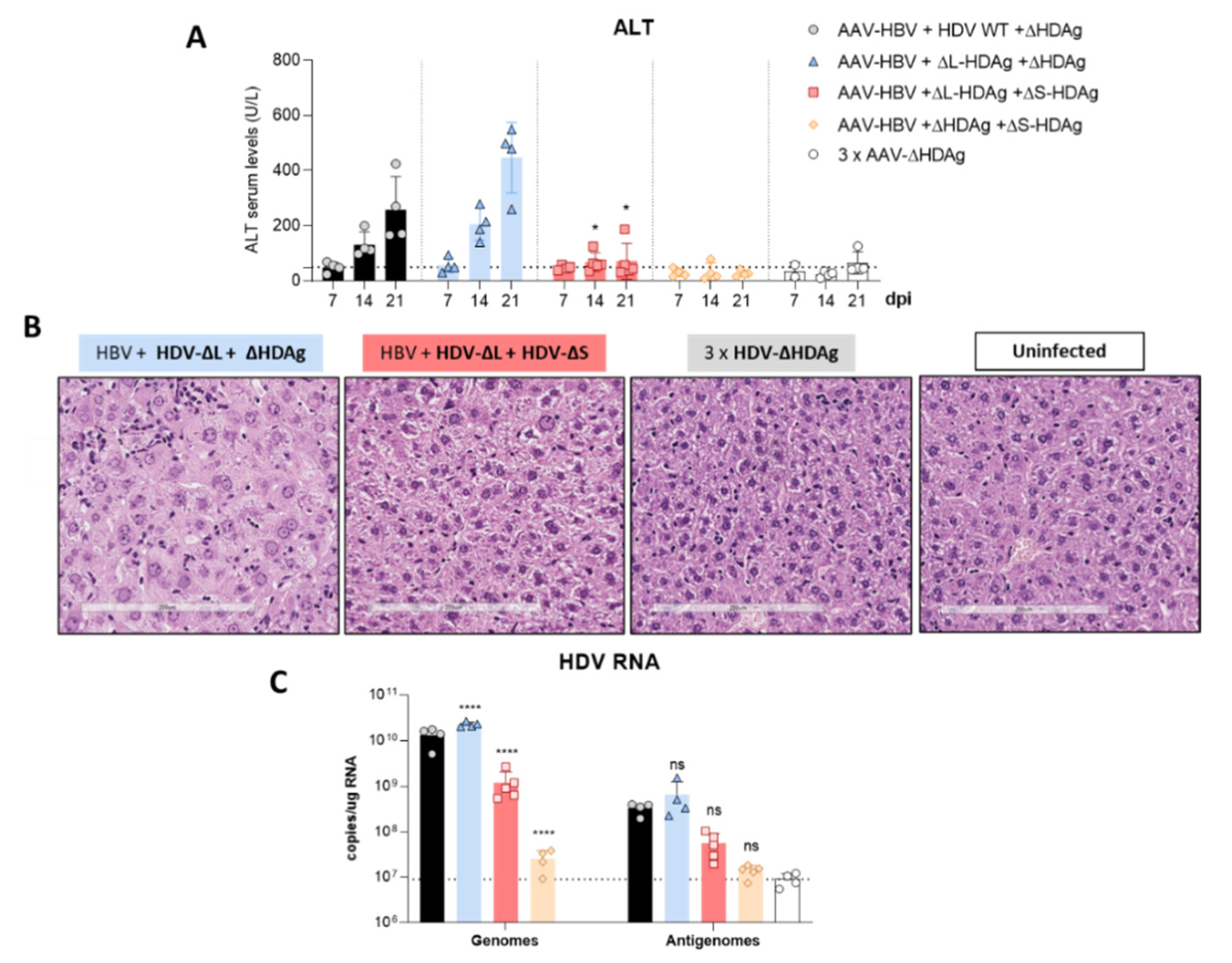
41]. The decrease in HDV replication from day 14 to day 21 observed in mice receiving the HDV-ΔL-HDAg mutant could be associated with higher levels of liver damage and apoptotic hepatocytes.
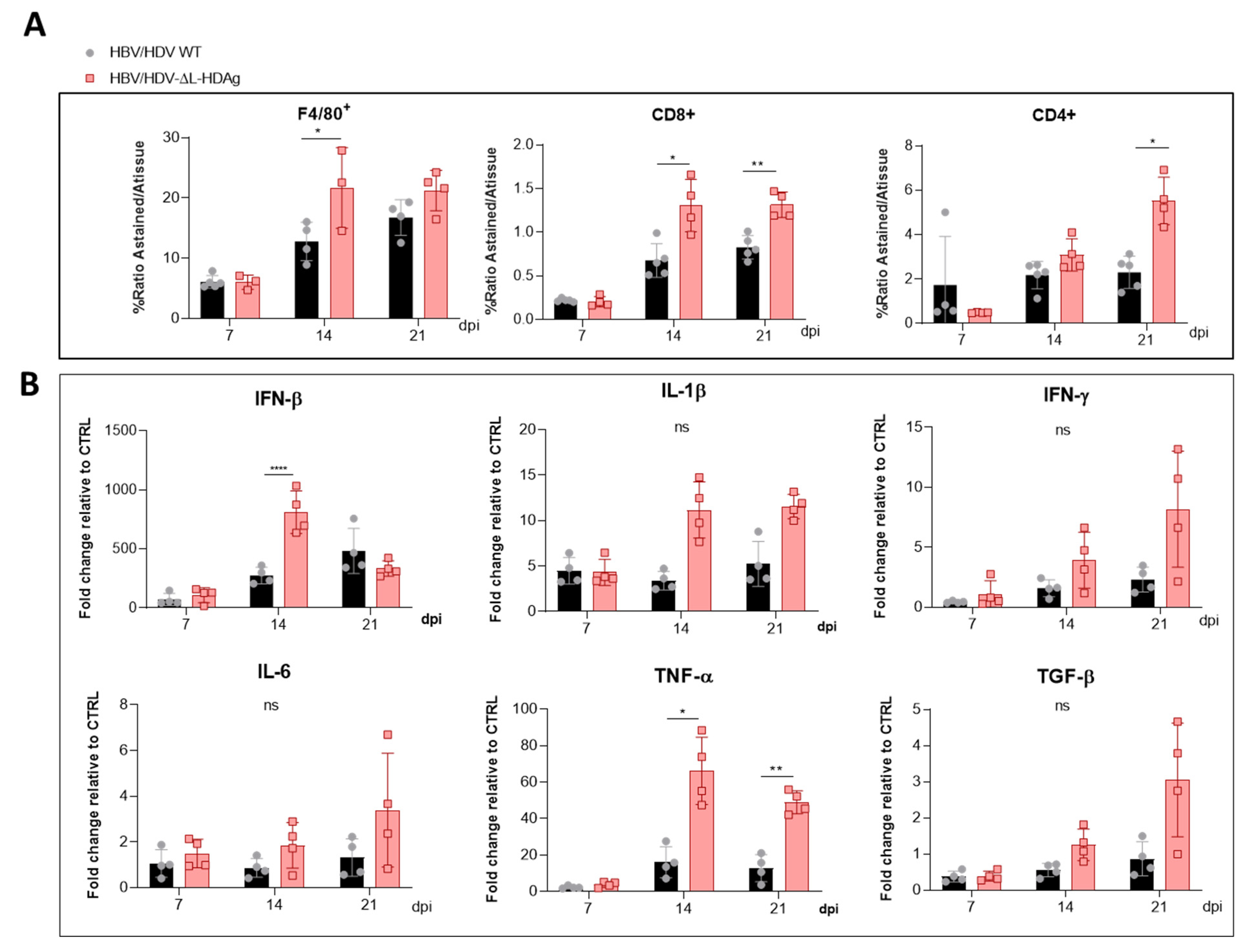
Furthermore, we found that production of the antigenome sequence alone or in the presence of L-HDAg had no effect on liver damage, while the expression of S-HDAg in the absence of L-HDAg resulted in a significant increase in liver damage in comparison with the damage induced by the injection of the AAV carrying the HDV WT genome. As shown, mice receiving the HDV-ΔL-HDAg mutant presented a higher number of aberrant hepatocytes with bigger nuclei as well as a higher frequency of apoptotic cells in comparison to HDV WT. Moreover, we also found that the liver of these animals showed a more prominent inflammatory infiltrate (with significantly higher numbers of macrophages and T cells), resulting in higher expression levels of TNF-α, which we previously showed to play a significant role in HDV-induced liver damage [
29]. Interestingly, the experiment performed in RagB6 mice, apart from demonstrating the essential role of L-HDAg in the formation of HDV infectious particles, also showed that T cells are not directly involved in the exacerbation of the liver disease induced by the HDV mutant expressing only S-HDAg, suggesting a direct cytotoxic effect associated with the overexpression and nuclear accumulation of S-HDAg. These results are in line with previous data demonstrating a direct cytotoxicity of S-HDAg [
13,
14,
15]. Cole et al. observed that cells stably transfected with a replication-competent HDV construct showed spontaneous death that diminished when the expression of L-HDAg was detected [
15]. Furthermore, it was reported that the L-HDAg/S-HDAg ratio is higher in patients with chronic HDV infection and low transaminase levels than in patients with HDV acute infection and high transaminase levels [
16], suggesting, as we observed here, a major role of S-HDAg in the development of HDV-induced liver damage that seems to be attenuated by L-HDAg expression. In fact, we found that the coadministration of the mutant expressing only L-HDAg and the mutant expressing only S-HDAg, which resulted in an early expression of both antigens after AAV administration, significantly ameliorated the liver damage induced by the mutant that only expresses S-HDAg. We also observed, as expected, that L-HDAg reduced both HDV replication and S-HDAg expression levels. With these experiments, we cannot discard that L-HDAg plays a role in HDV-induced pathology. Several studies have shown that the expression of L-HDAg may alter cellular homeostasis and induce liver damage. However, using the AAV-HDV delivery system, it is impossible to isolate the effect of L-HDAg from that of S-HDAg and HDV replication since HDV-ΔS-HDAg transiently expresses L-HDAg, and HDV replication is not initiated.
Additionally, our data indicate that hepatocyte infection with HDV viral particles carrying an edited genome will result in a sterile/unproductive infection since HDV replication cannot be initiated, and it will be productive only if the same cell is coinfected by an HDV carrying a non-edited genome. This is a very interesting finding considering that in patients, both non-edited and edited forms of the HDV genomes can be found in the circulation [
42,
43]. The role of the production of HDV particles containing an edited genome that will lead to a sterile/unproductive infection is not well understood. However, we might hypothesize that as the infection progresses, the viral particles harbouring edited genomes will enter cells in which HDV is actively replicating and will reduce viral replication and spread through the liver, but it will also ameliorate the activation of the HDV-induced inflammatory response and S-HDAg cytotoxicity. Using this strategy, the virus would avoid the death of infected cells and clearance by the immune system, improving the survival of the virus as well as the host cell. These findings are consistent with the hypothesis that L-HDAg may promote viral persistence by ameliorating liver damage [
15].
In summary, the use of AAV-HBV/AAV-HDV has allowed us to confirm in vivo the properties attributed to the HDAgs in the HDV life cycle previously described almost exclusively in cell culture. However, more importantly, we demonstrated the major role of HDAgs, mainly S-HDAg, in the HDV-induced liver pathology that was described in vitro in the early 90′s.
Additional experimentation is required to determine the reasons leading to HDV-associated liver damage observed for the first time in this mouse model, as opposed to others. Both the high efficacy of AAVs in delivering their genetic cargo into the hepatocytes and the strong liver-specific promoter controlling the synthesis of the initial copies of the antigenome are plausible explanations.
It is nonetheless clear that in this model, HDV replication and/or S-HDAg expression are required for the development of the observed liver pathology, since AAV vectors carrying either HBV replication-competent genomes or replication-deficient HDV genomes failed to induce liver damage.
Thus, AAV-mediated delivery of hepatitis virus replication-competent genomes represents a very attractive platform to determine the mechanism of HDV-induced liver pathology and for the development of new and more efficient treatments.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}