
Diversity and Reassortment Rate of Influenza A Viruses in Wild Ducks and Gulls



Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. Sequencing
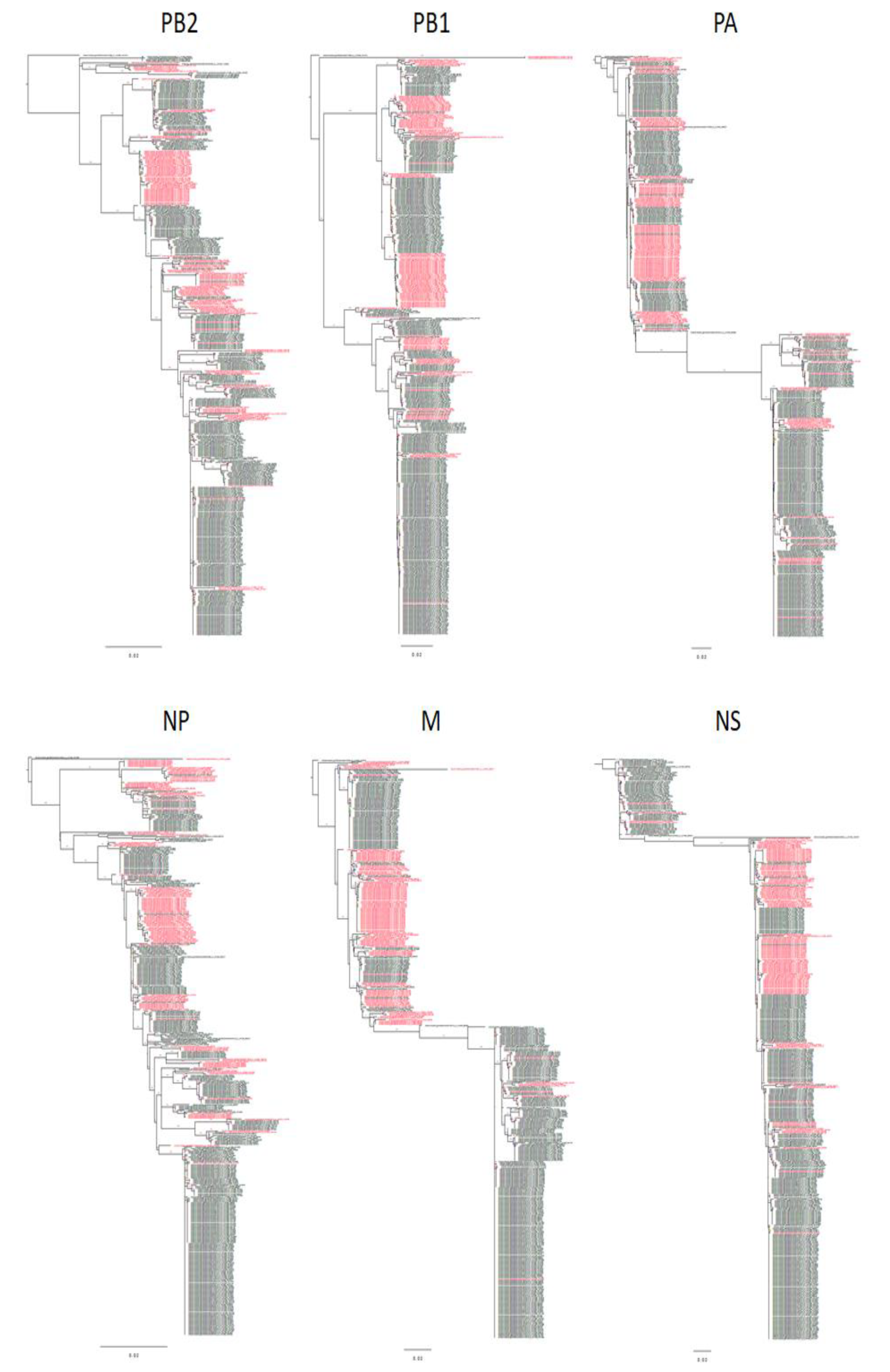
2.3. Downloading of Sequences and Evolutionary Tree Construction
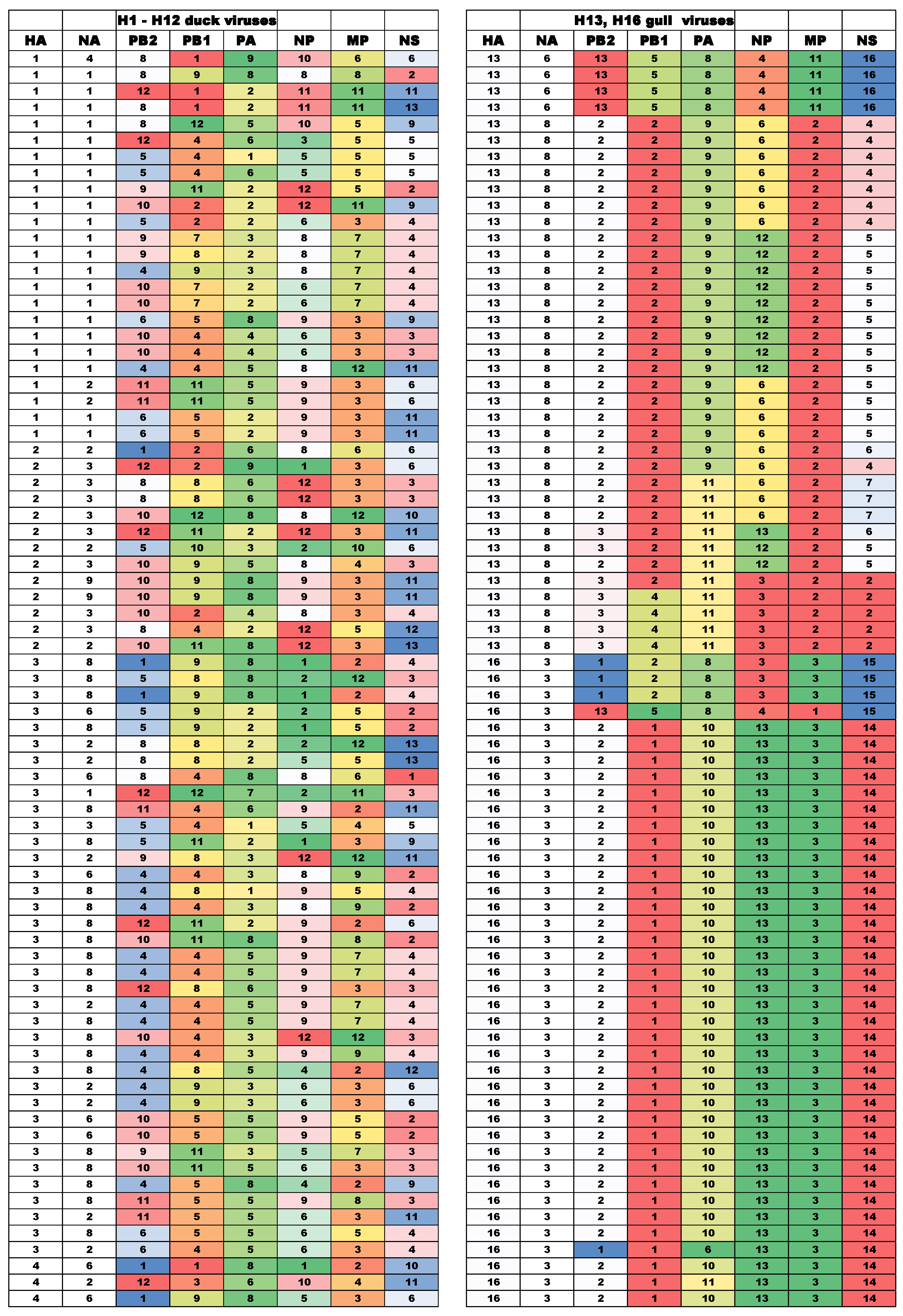
2.4. Classification of Gene Variants and Detection of Gene Reassortants
3. Results
3.1. Evolutionary Relationships of Gene Segments
3.2. Diversity of Gene Segment Constellations of IAVs in Ducks and Gulls
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza a virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munster, V.J.; Fouchier, R.A. Avian influenza virus: Of virus and bird ecology. Vaccine 2009, 45, 6340–6344. [Google Scholar] [CrossRef]
- Arnal, A.I.; Vittecoq, M.; Pearce-Duvet, J.; Gauthier-Clerc, M.; Boulinier, T.; Jourdain, E. Laridae: A neglected reservoir that could play a major role in avian influenza virus epidemiological dynamics. Crit. Rev. Microbiol. 2014, 41, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, J.H.; Majoor, F.; Lexmond, P.; Vuong, O.; Kasemir, G.; Lutterop, D.; Osterhaus, A.D.; Fouchier, R.A.; Kuiken, T. Epidemiology of influenza A virus among black-headed gulls, the Netherlands, 2006–2010. Emerging Infect. Dis. 2014, 1, 138–141. [Google Scholar] [CrossRef]
- Steel, J.; Lowen, A.C. Influenza A virus reassortment. Curr. Top Microbiol. Immunol. 2014, 385, 377–401. [Google Scholar] [CrossRef]
- Reid, A.H.; Taubenberger, J.K. The origin of the 1918 pandemic influenza virus: A continuing enigma. J. Gen. Virol. 2003, 84 Pt 9, 2285–2292. [Google Scholar] [CrossRef]
- Holmes, E.C.; Ghedin, E.; Miller, N.; Taylor, J.; Bao, Y.; St George, K.; Grenfell, B.T.; Salzberg, S.L.; Fraser, C.M.; Lipman, D.J.; et al. Whole-genome analysis of human influenza A virus reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biol. 2005, 9, e300. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Edelman, L.; Spiro, D.J.; Boyne, A.R.; Bera, J.; Halpin, R.; Ghedin, E.; Miller, M.A.; Simonsen, L.; Viboud, C.; et al. Molecular epidemiology of A/H3N2 and A/H1N1 influenza virus during a single epidemic season in the United States. PLoS Pathog. 2008, 4, e1000133. [Google Scholar] [CrossRef]
- Nelson, M.I.; Viboud, C.; Simonsen, L.; Bennett, R.T.; Griesemer, S.B.; George, K.S.; Taylor, J.; Spiro, D.J.; Sengamalay, N.A.; Ghedin, E.; et al. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog. 2008, 4, e1000012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Westgeest, K.B.; Russell, C.A.; Lin, X.; Spronken, M.I.; Bestebroer, T.M.; Bahl, J.; van Beek, R.; Skepner, E.; Halpin, R.A.; de Jong, J.C.; et al. Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J. Virol. 2014, 88, 2844–2857. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, F.; Sugiyama, T. Direct isolation of H1N2 recombinant virus from a throat swab of a patient simultaneously infected with H1N1 and H3N2 influenza A viruses. J. Clin. Microbiol. 1983, 18, 425–427. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, T.R.; Agrawal, A.S.; Chakrabarti, S.; Chawla-Sarkar, M. Full genomic analysis of an influenza A (H1N2) virus identified during 2009 pandemic in Eastern India: Evidence of reassortment event between co-circulating A(H1N1)pdm09 and A/Brisbane/10/2007-like H3N2 strains. Virol. J. 2012, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; La, T.; Zhao, P.; Tam, J.S.; Rappaport, R.; Cheng, S.M. Genetic and phylogenetic analysis of multi-continent human influenza A(H1N2) reassortant viruses isolated in 2001 through 2003. Virus Res. 2006, 122, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Al Faress, S.; Ferraris, O.; Moules, V.; Valette, M.; Hay, A.; Lina, B. Identification and characterization of a late AH1N2 human reassortant in France during the 2002–2003 influenza season. Virus Res. 2008, 132, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Neverov, A.D.; Lezhnina, K.V.; Kondrashov, A.S.; Bazykin, G.A. Intrasubtype reassortments cause adaptive amino acid replacements in H3N2 influenza genes. PLoS Genet. 2014, 10, e1004037. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.; Lässig, M. Fitness cost of reassortment in human influenza. PLoS Pathog. 2017, 13, e1006685. [Google Scholar] [CrossRef] [Green Version]
- Phipps, K.L.; Marshall, N.; Tao, H.; Danzy, S.; Onuoha, N.; Steel, J.; Lowen, A.C. Seasonal H3N2 and 2009 Pandemic H1N1 Influenza A Viruses Reassort Efficiently but Produce Attenuated Progeny. J. Virol. 2017, 91, e00830-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholtissek, C. Molecular evolution of influenza viruses. Virus Genes 1995, 11, 209. [Google Scholar] [CrossRef]
- Karasin, A.I.; Schutten, M.M.; Cooper, L.A.; Smith, C.B.; Subbarao, K.; Anderson, G.A.; Carman, S.; Olsen, C.W. Genetic characterization of H3N2 influenza viruses isolated from pigs in North America; 1977–1999: Evidence for wholly human and reassortant virus genotypes. Virus Res. 2000, 68, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Karasin, A.I.; Carman, S.; Olsen, C.W. Identification of human H1N2 and human-swine reassortant H1N2 and H1N1 influenza A viruses among pigs in Ontario; Canada (2003 to 2005). J. Clin. Microbiol. 2006, 44, 1123–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobolev, I.; Kurskaya, O.; Leonov, S.; Kabilov, M.; Alikina, T.; Alekseev, A.; Yushkov, Y.; Saito, T.; Uchida, Y.; Mine, J.; et al. Novel reassortant of H1N1 swine influenza virus detected in pig population in Russia. Emerg. Microbes Infect. 2019, 8, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Zhang, P.C.; Zhou, Y.J.; Li, G.X.; Pan, J.; Yan, L.P.; Shi, X.X.; Liu, H.L.; Tong, G.Z. Isolation and genetic characterization of avian-like H1N1 and novel ressortant H1N2 influenza viruses from pigs in China. Biochem. Biophys Res. Commun. 2009, 386, 278–283. [Google Scholar] [CrossRef]
- Nardelli, L.; Pascucci, S.; Gualandi, G.L.; Loda, P. Outbreaks of classical swine influenza in Italy in 1976. Zent. Vet. B 1978, 25, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.M.; Hinshaw, V.S.; Kawaoka, Y.; Easterday, B.C.; Webster, R.G. Influenza viral infection of swine in the United States 1988–1989. Arch. Virol. 1991, 116, 261–265. [Google Scholar] [CrossRef]
- Worobey, M.; Han, G.Z.; Rambaut, A. A synchronized global sweep of the internal genes of modernavian influenza virus. Nature 2014, 508, 254–257. [Google Scholar] [CrossRef]
- Lin, Y.P.; Shaw, M.; Gregory, V.; Cameron, K.; Lim, W.; Klimov, A.; Subbarao, K.; Guan, Y.; Krauss, S.; Shortridge, K.; et al. Avian-to-human transmission of H9N2 subtype influenza A viruses: Relationship between H9N2 and H5N1 human isolates. Proc. Natl. Acad. Sci. USA 2000, 97, 9654–9658. [Google Scholar] [CrossRef] [Green Version]
- White, M.C.; Lowen, A.C. Implications of segment mismatch for influenza A virus evolution. J. Gen. Virol. 2018, 99, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, A.; Mahmoud, S.H.; Shehata, M.; Müller, C.; Kandeil, A.; El-Shesheny, R.; Nooh, H.Z.; Kayali, G.; Ali, M.A.; Pleschka, S. PA from a Recent H9N2 (G1-Like) Avian Influenza a Virus (AIV) Strain Carrying Lysine 367 Confers Altered Replication Efficiency and Pathogenicity to Contemporaneous H5N1 in Mammalian Systems. Viruses 2020, 12, 1046. [Google Scholar] [CrossRef]
- Feare, C.J. The role of wild birds in the spread of HPAI H5N1. Avian Dis. 2007, 51, 440–447. [Google Scholar] [CrossRef]
- Fan, S.; Zhou, L.; Wu, D.; Gao, X.; Pei, E.; Wang, T.; Gao, Y.; Xia, X. A novel highly pathogenic H5N8 avian influenza virus isolated from a wild duck in China. Influenza Other Respi. Viruses 2014, 8, 646–653. [Google Scholar] [CrossRef]
- Krauss, S.; Stallknecht, D.E.; Slemons, R.D.; Bowman, A.S.; Poulson, R.L.; Nolting, J.M.; Knowles, J.P.; Webster, R.G. The enigma of the apparent disappearance of Eurasian highly pathogenic H5 clade 2.3.4.4 influenza A viruses in North American waterfowl. Proc. Natl. Acad. Sci. USA 2016, 113, 9033–9038. [Google Scholar] [CrossRef] [Green Version]
- Dugan, V.G.; Chen, R.; Spiro, D.J.; Sengamalay, N.; Zaborsky, J.; Ghedin, E.; Nolting, J.; Swayne, D.E.; Runstadler, J.A.; Happ, G.M.; et al. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLoS Pathog. 2008, 4, e1000076. [Google Scholar] [CrossRef] [Green Version]
- Chambers, T.M.; Yamnikova, S.; Kawaoka, Y.; Lvov, D.K.; Webster, R.G. Antigenic and molecular characterization of subtype H13 hemagglutinin of influenza virus. Virology 1989, 172, 180–188. [Google Scholar] [CrossRef]
- Fouchier, R.A.; Munster, V.; Wallensten, A.; Bestebroer, T.M.; Herfst, S.; Smith, D.; Rimmelzwaan, G.F.; Olsen, B.; Osterhaus, A.D. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol. 2005, 79, 2814–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, M.; Holmes, E.C. The Ecology and Evolution of Influenza Viruses. Cold Spring Harb. Perspect. Med. 2020, 10, a038489. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.S.; Hallgrimsson, G.T.; Suwannanarn, K.; Sreevatsen, S.; Ip, H.S.; Magnusdottir, E.; TeSlaa, J.L.; Nashold, S.W.; Dusek, R.J. Avian influenza virus ecology in Iceland shorebirds: Intercontinental reassortment and movement. Infect. Genet. Evol. 2014, 28, 130–136. [Google Scholar] [CrossRef]
- Hall, J.S.; Teslaa, J.L.; Nashold, S.W.; Halpin, R.A.; Stockwell, T.; Wentworth, D.E.; Dugan, V.; Ip, H.S. Evolution of a reassortant North American gull influenza virus lineage: Drift; shift and stability. Virol. J. 2013, 10, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhagen, J.H.; Poen, M.; Stallknecht, D.E.; van der Vliet, S.; Lexmond, P.; Sreevatsan, S.; Poulson, R.L.; Fouchier, R.A.M.; Lebarbenchon, C. Phylogeography and Antigenic Diversity of Low-Pathogenic Avian Influenza H13 and H16 Viruses. J. Virol. 2020, 94, e00537-20. [Google Scholar] [CrossRef]
- Marshall, N.; Priyamvada, L.; Ende, Z.; Steel, J.; Lowen, A.C. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog. 2013, 9, e1003421. [Google Scholar] [CrossRef] [Green Version]
- Fonville, J.M.; Marshall, N.; Tao, H.; Steel, J.; Lowen, A.C. Influenza Virus Reassortment Is Enhanced by Semi-infectious Particles but Can Be Suppressed by Defective Interfering Particles. PLoS Pathog. 2015, 11, e1005204. [Google Scholar] [CrossRef]
- Tao, H.; Li, L.; White, M.C.; Steel, J.; Lowen, A.C. Influenza A virus coinfection through transmission can support high levels of reassortment. J. Virol. 2015, 89, 8453–8461. [Google Scholar] [CrossRef] [Green Version]
- Li, O.T.; Barr, I.; Leung, C.Y.; Chen, H.; Guan, Y.; Peiris, J.S.; Poon, L.L. Reliable universal RT-PCR assays for studying influenza polymerase subunit gene sequences from all 16 haemagglutinin subtypes. J. Virol. Methods 2007, 142, 218–222. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Q.; Wang, S.; Liu, S.; Hou, G.; Li, J.; Jiang, W.; Wang, K.; Peng, C.; Liu, D.; Guo, A.; et al. Diversity and distribution of type A influenza viruses: An updated panorama analysis based on protein sequences. Virol. J. 2019, 16, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boravleva, E.Y.; Lomakina, N.F.; Gambaryan, A.S. Isolation of influenza A viruses from birds on ponds of Moscow. Kazarka 2012, 15, 13–30. (In Russian) [Google Scholar]
- Heydarov, R.N.; Lomakina, N.F.; Boravleva, E.Y.; Kholodilov, I.S.; Gambaryan, A.S.; Mikhailovich, V.M.; Fesenko, E.E. The use of microarrays for the identification of the origin of genes of avian influenza viruses in wild birds. MIR J. 2017, 4, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, J.H.; van der Jeugd, H.P.; Nolet, B.A.; Slaterus, R.; Kharitonov, S.P.; de Vries, P.P.; Vuong, O.; Majoor, F.; Kuiken, T.; Fouchier, R.A. Wild bird surveillance around outbreaks of highly pathogenic avian influenza A(H5N8) virus in the Netherlands; 2014; within the context of global flyways. Eurosurveillance 2015, 20, 21069. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Poen, M.J.; Bestebroer, T.M.; Scheuer, R.D.; Vuong, O.; Chkhaidze, M.; Machablishvili, A.; Mamuchadze, J.; Ninua, L.; Fedorova, N.B.; et al. Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem. J. Virol. 2018, 92, e00433-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, M.; Tolf, C.; Avril, A.; Latorre-Margalef, N.; Wallerström, S.; Olsen, B.; Waldenström, J. Frequency and patterns of reassortment in natural influenza A virus infection in a reservoir host. J. Virol. 2013, 443, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Swayne, D.E. Understanding the complex pathobiology of high pathogenicity avian influenza viruses in birds. Avian Dis. 2007, 51 (Suppl. 1), 242–249. [Google Scholar] [CrossRef]
- Munster, V.J.; Schrauwen, E.J.; de Wit, E.; van den Brand, J.M.; Bestebroer, T.M.; Herfst, S.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Insertion of a multibasic cleavage motif into the hemagglutinin of a low-pathogenic avian influenza H6N1 virus induces a highly pathogenic phenotype. J. Virol. 2010, 84, 7953–7960. [Google Scholar] [CrossRef] [Green Version]
- Suguitan, A.L., Jr.; Matsuoka, Y.; Lau, Y.F.; Santos, C.P.; Vogel, L.; Cheng, L.I.; Orandle, M.; Subbarao, K. The multibasic cleavage site of the hemagglutinin of highly pathogenic A/Vietnam/1203/2004 (H5N1) avian influenza virus acts as a virulence factor in a host-specific manner in mammals. J. Virol. 2012, 8, 2706–2714. [Google Scholar] [CrossRef] [Green Version]
- Kaverin, N.V.; Gambaryan, A.S.; Bovin, N.V.; Rudneva, I.A.; Shilov, A.A.; Khodova, O.M.; Varich, N.L.; Sinitsin, B.V.; Makarova, N.V.; Kropotkina, E.A. Postreassortment changes in influenza A virus hemagglutinin restoring HA-NA functional match. Virology 1998, 244, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambaryan, A.S.; Matrosovich, T.Y.; Boravleva, E.Y.; Lomakina, N.F.; Yamnikova, S.S.; Tuzikov, A.B.; Pazynina, G.V.; Bovin, N.V.; Fouchier, R.A.M.; Klenk, H.D.; et al. Receptor-binding properties of influenza viruses isolated from gulls. Virology 2018, 522, 37–45. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Isolation Date | Strain Name a | Subtype | Clade/Subclade b | |||||
|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | NP | M | NS | |||
| 15 October 2019 | M/5743/2019 | H1N1 | 6 | 5 | 2a | 10b | 4E | B11 |
| 15 October 2019 | M/5744/2019 | H1N1 | 6 | 5 | 2a | 10b | 4E | B11 |
| 1 October 2013 | M/4970/2013 | H1N1 | 6 | 5 | 8a | 10c | 4D | B9d |
| 17 October 2018 | M/5586/2018 | H1N2 | 11 | 12A | 5a | 10b | 4E | 6a |
| 1 November 2018 | M/5662/2018 | H1N2 | 11 | 12A | 5a | 10b | 4E | 6a |
| 13 October 2008 | M/4203/2010 | H3N8 | 4 | 4C | 3a | 9B | 10 | 2b |
| 26 November 2010 | M/4238/2010 | H3N6 | 4 | 4C | 3a | 9B | 10 | 2b |
| 13 October 2010 | M/4298/2010 | H3N8 | 4 | 9C | 1 | 10e | 5e | 4d |
| 27 September 2011 | M/4494/2011 | H3N8 | 4 | 4A | 5b | 10d | 7 | 4c |
| 10 October 2011 | M/4681/2011 | H3N8 | 4 | 4A | 5b | 10d | 7 | 4c |
| 1 October 2011 | M/4524/2011mix | H3N2 | 4 | 4C | 5b | 10d | 7 | 4c |
| 1 October 2011 | M/4524/2011mix | H3N8 | 4 | 4C | 5b | 10d | 7 | 4c |
| 1 October 2012 | M/4780/2012 | H3N8 | 4 | 9C | 5a | 4 | 3 | B12 |
| 1 October 2012 | M/4788/2012 | H3N8 | 4 | 4C | 3a | 10d | 10 | 4c |
| 4 September 2009 | M/3806/2009 | H3N8 | 11 | 4C | 6b | 10d | 3 | B11 |
| 1 October 2014 | M/5037/2014 | H3N8 | 11 | 5 | 5b | 10a | 9 | 3c |
| 13 October 2008 | M/3556/2008 | H3N1 | 12 | 13 | 7 | 2B | 12 | 3d |
| 13 November 2010 | M/4242/2010 | H3N8 | 12 | 12A | 2b | 10d | 3 | 6a |
| 10 November 2011 | M/4661/2011 | H3N8 | 12 | 9C | 6b | 10d | 4E | 3e |
| 19 September 2009 | M/3740/2009 | H4N6 | 2 | 9C | 1 | 10e | 5e | B11 |
| 4 September 2009 | M/3799/2009 | H4N6 | 2 | 9C | 1 | 10e | 1 | B11 |
| 19 September 2009 | M/3735/2009 | H4N6 | 3 | 9C | 1 | 10d | 1 | B11 |
| 31 October 2012 | M/4781/2012 | H4N6 | 4 | 4C | 3a | 10d | 7 | 3e |
| 17 October 2012 | M/4843/2012 | H4N6 | 4 | 4C | 3a | 3 | 7 | 4c |
| 10 October 2012 | M/4771/2012 | H4N6 | 6 | 4A | 3a | 3 | 7 | 4c |
| 11 October 2011 | M/4643/2011 | H4N6 | 10 | 10 | 8b | 9B | 4E | 2c |
| 21 October 2008 | M/3661/2008 | H4N6 | 12 | 13 | 7 | 12 | 6 | 3d |
| 4 October 2011 | M/4518/2011 | H4N6 | 12 | 9B | 5a | 9B | 5e | 2c |
| 4 October 2011 | M/4528/2011 | H4N6 | 12 | 4C | 5a | 10d | 5e | 4c |
| 19 October 2011 | M/4641/2011 | H4N6 | 12 | 4C | 5a | 10d | 5e | 4c |
| 16 November 2010 | M/4182/2010 | H5N3 | 11 | 4A | 2a | 10d | 3 | 3d |
| 19 November 2013 | M/4971/2013 | H5N3 | 4 | 4C | 6b | 10d | 10 | 2b |
| 26 November 2013 | M/4952/2013 | H5N3 | 11 | 4A | 5b | 10b | 6 | 2b |
| 1 September 2009 | M/3720/2009 | H6N2 | 6 | 4A | 2b | 3 | 12 | B11 |
| 1 September 2010 | M/4031/2010 | H6N2 | 12 | 4A | 2a | 13 | 13 | 3c |
| 11 October 2006 | gull/M/3100/2006 | H6N2 | 1c | 10 | 7 | 1 | 6 | 6b |
| 4 November 2008 | M/3641/2008 | H11N9 | 12 | 9A | 3b | 5 | 11 | 3d |
| 21 October 2019 | M/5712/2019 | H11N6 | 11 | 12A | 5a | 10b | 4E | 3e |
| Duck Viruses Isolated in Moscow and The Netherlands | Gull Viruses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| H1 | H2 | H3 | H4 | H5 | H6 | H11 | H13 | H16N3 | |
| NI | 24 | 13 | 37 | 31 | 19 | 34 | 9 | 252 | 94 |
| NG | 20 | 11 | 34 | 29 | 18 | 29 | 9 | 84 | 38 |
| % | 83 | 85 | 92 | 94 | 95 | 85 | 100 | 33 | 40 |
| Strain Number | Clade/Subclade a | Strain Number | Clade/Subclade | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | NP | MP | NS | PB2 | PB1 | PA | NP | MP | NS | ||
| bhg/N/xx/2007|H13N6 b | bhg/N/xx/2007|H16N3 | ||||||||||||
| 2 | 13 | 5 | 8 | 14 | 11 | 16a | 8 | 1 | 2b | 8 | 3 | 3f | 15 |
| 4 | 13 | 5 | 8 | 14 | 11 | 16a | 3 | 1 | 2b | 8 | 3 | 3f | 15 |
| 6 | 13 | 5 | 8 | 14 | 11 | 16a | 9 | 1 | 2b | 8 | 3 | 3f | 15 |
| 10 | 13 | 5 | 8 | 14 | 11 | 16a | 7 | 13 | 5 | 8 | 4 | 1 | 15 |
| bhg/N/xx/2008|H13N8 | bhg/N/xx/2008|H16N3 | ||||||||||||
| 15 | 2b | 2a | 9 | 6 | 2b | 6 | 84 | 2a | 1a | 10b | 13 | 3a | 14a |
| 55 | 2b | 2a | 9 | 6 | 2b | 4 | 56 | 2a | 1a | 10b | 13 | 3a | 14a |
| 51 | 3c | 2a | 11b | 13 | 2b | 6 | 33 | 1 | 1a | 8 | 13 | 3a | 14a |
| 52 | 3c | 2a | 9 | 13 | 2b | 6 | 92 | 2a | 1a | 10b | 13 | 3b | 14a |
| bhg/N/xx/2009|H13N2, N6, N3 | bhg/N/xx/2009|H16N3 | ||||||||||||
| 27 (N2) | 11a | 3d | 10a | 3 | 11 | 2c | 35 | 11c | 3d | 10b | 7 | 3b | 2e |
| 38 (N2) | 3a | 1b | 10b | 7 | 5 | 16b | 25 | 11c | 3d | 10b | 15 | 3b | 2e |
| 31 (N2) | 12 | 6b | 6a | 2 | 10 | 16b | 21 | 11c | 3d | 10b | 15 | 3b | 2e |
| 39 (N6) | 12 | 6b | 6a | 2 | 10 | 16b | 28 | 11c | 3d | 10b | 15 | 3b | 2e |
| 17 (N3) | 11a | 6b | 11c | 11 | 10 | 16b | 23 | 11a | 3a | 11c | 11 | 3c | 2c |
| bhg/N/xx/2011|H13N8, N3 | 22 | 11c | 3b | 10c | 15 | 3b | 2e | ||||||
| 19 | 11c | 3c | 10c | 15 | 7c | 11 | bhg/N/xx/2011|H16N3 | ||||||
| 9 | 11c | 3c | 10c | 15 | 7c | 11 | 28 | 11c | 3c | 10c | 9 | 3b | 2e |
| 39 | 11c | 3c | 10c | 15 | 3d | 8 | bhg/N/xx/2011|H16N3 | ||||||
| 8 (N3) | 11b | 10 | 10a | 15 | 3d | 2c | 33 | 11b | 10 | 10a | 15 | 3d | 2c |
| bhg/N/xx/2012|H13N6 | 30 | 11b | 10 | 10a | 15 | 3d | 2c | ||||||
| 96 | 8a | 8e | 3b | 8d | 8e | 9a | 34 | 11b | 10 | 10a | 15 | 3d | 2c |
| 97 | 8a | 8e | 3b | 8d | 8e | 9a | 27 | 11b | 10 | 10a | 15 | 3d | 2c |
| bhg/N/xx/2014|H13N6 | 1 | 11b | 10 | 10c | 15 | 3d | 2c | ||||||
| 11 | 8c | 9b | 6b | 8c | 3c | 10d | 31 | 11b | 10c | 10a | 15 | 3d | 2c |
| 15 | 8c | 9b | 6b | 8c | 3c | 10d | bhg/N/xx/2012|H16N3 | ||||||
| 28 | 8c | 9b | 6b | 8c | 3c | 10d | 107 | 10 | 3c | 6d | 15 | 3c | 2c |
| 3 | 8c | 9b | 6b | 8c | 3c | 10d | 114 | 10 | 3c | 6d | 15 | 3c | 2c |
| 32 | 8c | 9b | 6b | 8c | 3c | 10d | bhg/N/xx/2013|H16N3 | ||||||
| 16 | 10 | 9b | 6b | 8c | 3c | 10d | 5 | 7c | 8a | 3c | 8b | 3d | 9a |
| bhg/N/xx/2014|H13N2 | 2 | 7c | 8a | 3c | 8b | 3d | 9a | ||||||
| 2 | 5 | 9a | 5a | 8c | 6 | 9a | 3 | 7c | 8a | 3c | 8b | 3d | 9a |
| 6 | 5 | 9a | 5a | 8c | 6 | 9a | 4 | 7c | 8a | 3c | 8b | 3d | 12 |
| 37 | 5 | 9a | 4 | 8a | 6 | 16b | bhg/N/xx/2015|H16N3 | ||||||
| 24 | 5 | 9a | 4 | 8a | 6 | 16b | 1 | 7c | 9b | 5c | 8c | 3d | 9a |
| bhg/N/xx/2016|H13N2 | bhg/N/xx/2016|H16N3 | ||||||||||||
| 11 | 9 | 8a | 5b | 8c | 7b | 16b | 1 | 5 | 12 | 3b | 8a | 3d | 9a |
| 2 | 9 | 8a | 5b | 8c | 7b | 16b | 3 | 5 | 12 | 3b | 8a | 3d | 9a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postnikova, Y.; Treshchalina, A.; Boravleva, E.; Gambaryan, A.; Ishmukhametov, A.; Matrosovich, M.; Fouchier, R.A.M.; Sadykova, G.; Prilipov, A.; Lomakina, N. Diversity and Reassortment Rate of Influenza A Viruses in Wild Ducks and Gulls. Viruses 2021, 13, 1010. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061010
Postnikova Y, Treshchalina A, Boravleva E, Gambaryan A, Ishmukhametov A, Matrosovich M, Fouchier RAM, Sadykova G, Prilipov A, Lomakina N. Diversity and Reassortment Rate of Influenza A Viruses in Wild Ducks and Gulls. Viruses. 2021; 13(6):1010. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061010
Chicago/Turabian StylePostnikova, Yulia, Anastasia Treshchalina, Elizaveta Boravleva, Alexandra Gambaryan, Aydar Ishmukhametov, Mikhail Matrosovich, Ron A. M. Fouchier, Galina Sadykova, Alexey Prilipov, and Natalia Lomakina. 2021. "Diversity and Reassortment Rate of Influenza A Viruses in Wild Ducks and Gulls" Viruses 13, no. 6: 1010. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061010