
An MHV-68 Mutator Phenotype Mutant Virus, Confirmed by CRISPR/Cas9-Mediated Gene Editing of the Viral DNA Polymerase Gene, Shows Reduced Viral Fitness

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Compounds
2.3. Selection of Drug-Resistant MHV-68
2.4. Genotypic Analysis
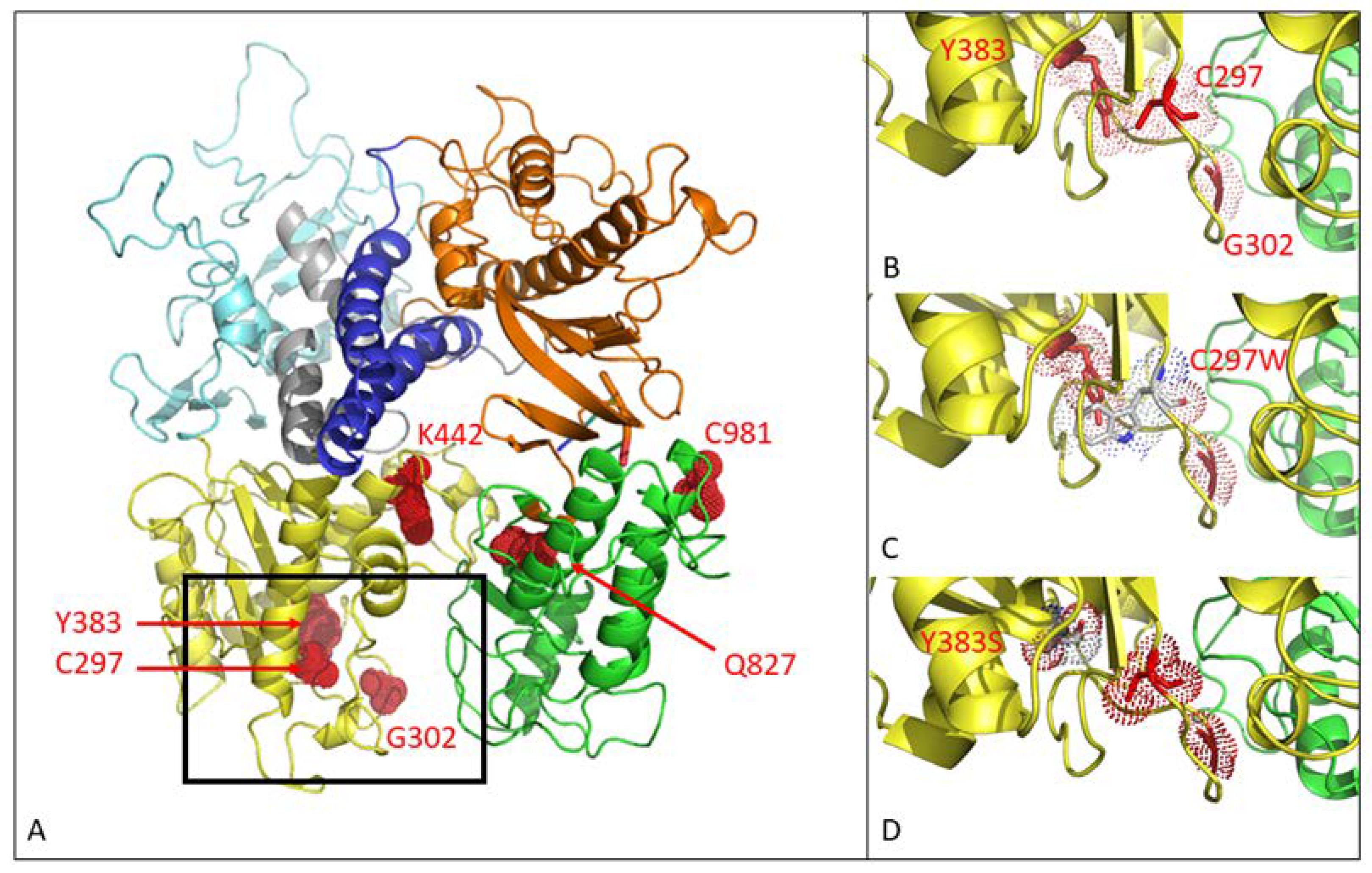
2.5. 3D Modeling
2.6. Drug Susceptibility Profile
2.7. Replication Capacity and Relative Fitness
2.8. Correlation between Antiviral Resistance and Relative Fitness
2.9. Investigation of the Mutation Frequency in the Viral PK, TK and DP
2.10. CRISPR/Cas9 Genome Editing of MHV-68 DP Amino Acid Position C297
2.10.1. Plasmid Construct
2.10.2. Transfection/Infection
3. Results
3.1. MHV-68 Mutants Selected under Pressure of GCV-, HPMP-5azaC- or PFA-Harbored Mutations in the Viral DNA Polymerase (DP)
3.2. MHV-68 Mutants Selected under Pressure of CDV or HPMPO-DAPy Were Associated with a Mutator Phenotype Virus
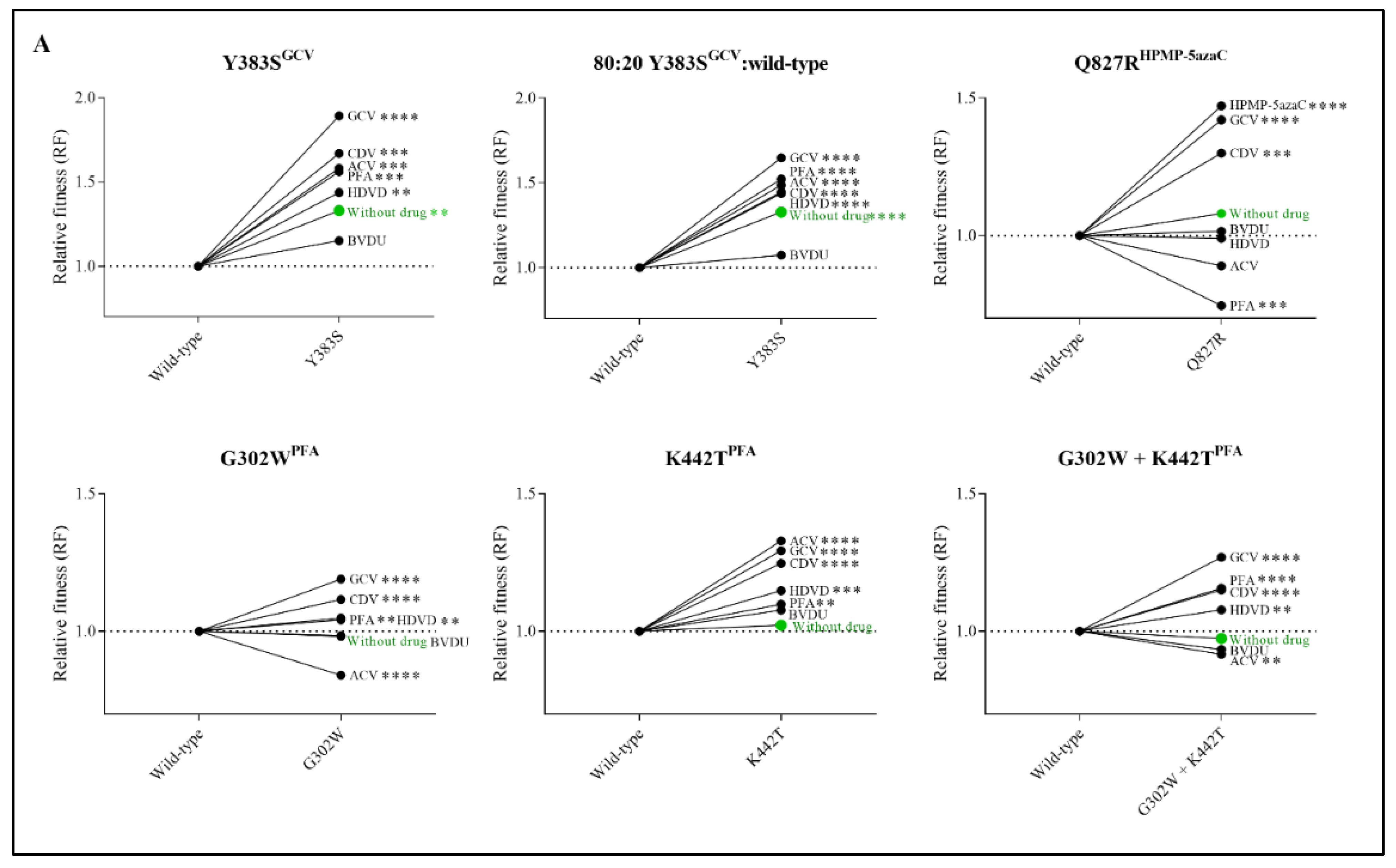
3.3. Without Antiviral Pressure, the Viral Fitness of Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA Remained Equal to Wild-Type Virus, While Y383SGCV Had an Increased Viral Fitness
3.4. Antiviral Drug Treatment Altered the Viral Fitness of Y383SGCV, Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA
3.5. Relative Fitness Strongly Correlated with Drug Resistance Levels for Y383SGCV, Q827RHPMP-5azaC, G302WPFA and G302W+K442TPFA, and Moderately for K442TPFA
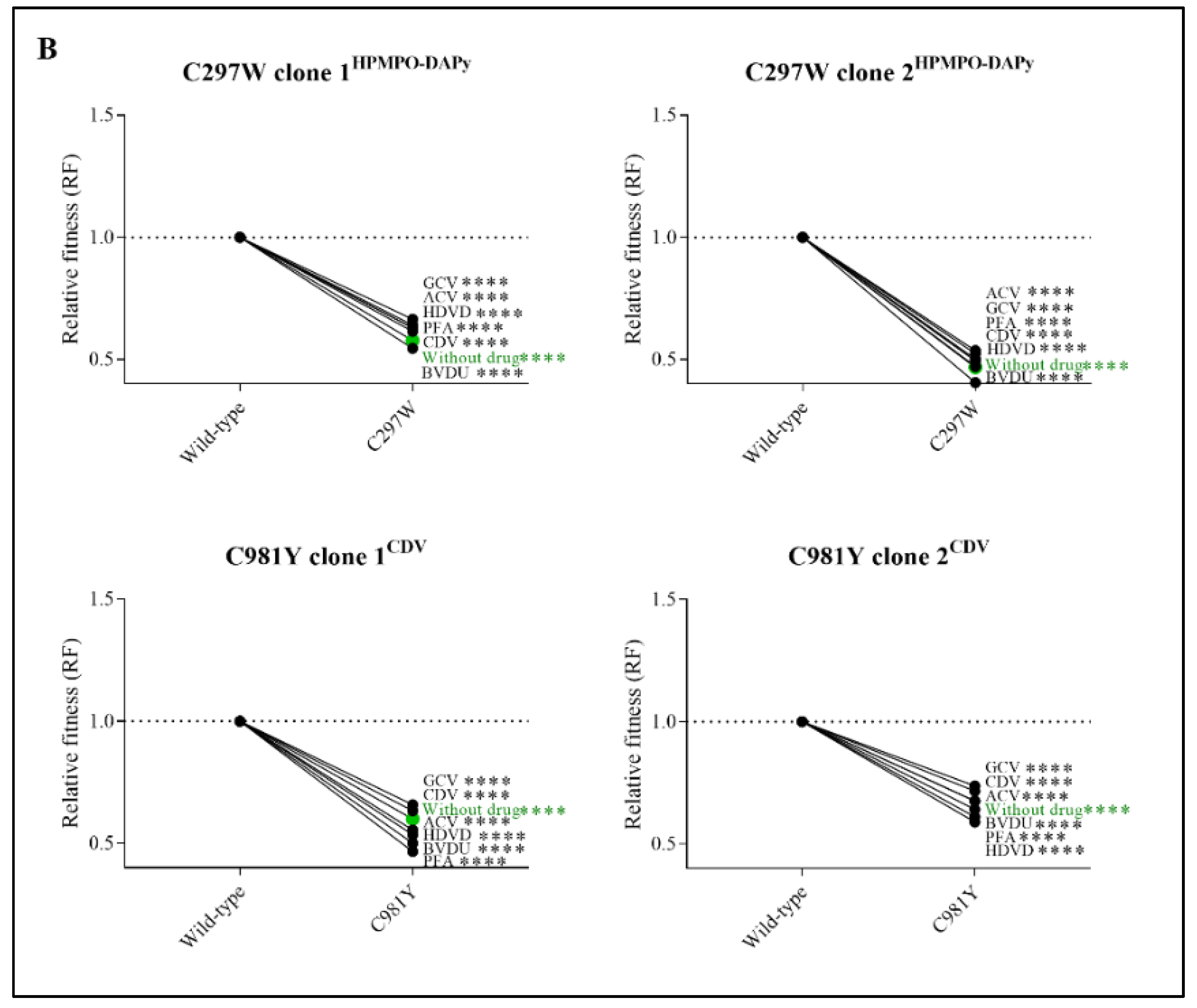
3.6. Mutator Phenotype Viruses Had a Severely Impaired Viral Fitness in the Absence and Presence of Antivirals
3.7. Mutator Phenotype Virus C981YCDV, but Not C297WHPMPO-DAPy, Demonstrated a Correlation between Relative Fitness and Antiviral Drug Resistance Levels
3.8. Investigation of the Mutation Frequency in the Viral PK, TK and DP
3.9. CRISPR/Cas9 Genome Editing Was a Successful Strategy for the Generation of MHV-68 Virus with a Missense Mutation Resulting in a Mutator Phenotype Virus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Distribution and effects of amino acid changes in drug-resistant alpha and beta herpesviruses DNA polymerase. Nucleic Acids Res. 2016, 44, 9530–9554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, D.W.; Szpara, M.L. Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Cesarman, E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011, 305, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, H.C.; Banerjee, S.; Robertson, E.S. The Role of Gammaherpesviruses in Cancer Pathogenesis. Pathogens 2016, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein-Barr Virus-Associated Malignancies: Roles of Viral Oncoproteins in Carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—An update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Manners, O.; Murphy, J.C.; Coleman, A.; Hughes, D.J.; Whitehouse, A. Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr. Opin. Virol. 2018, 32, 60–70. [Google Scholar] [CrossRef]
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus lytic cycle involvement in diffuse large B cell lymphoma. Hematol. Oncol. 2018, 36, 98–103. [Google Scholar] [CrossRef]
- Jones, R.J.; Seaman, W.T.; Feng, W.H.; Barlow, E.; Dickerson, S.; Delecluse, H.J.; Kenney, S.C. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int. J. Cancer 2007, 121, 1274–1281. [Google Scholar] [CrossRef]
- Beatty, P.R.; Krams, S.M.; Martinez, O.M. Involvement of IL-10 in the autonomous growth of EBV-transformed B cell lines. J. Immunol. 1997, 158, 4045–4051. [Google Scholar] [PubMed]
- Jones, K.D.; Aoki, Y.; Chang, Y.; Moore, P.S.; Yarchoan, R.; Tosato, G. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi’s sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 1999, 94, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, P.; Sakakibara, S.; Tosato, G. Contribution of viral and cellular cytokines to Kaposi’s sarcoma-associated herpesvirus pathogenesis. J. Leukoc Biol. 2008, 84, 994–1000. [Google Scholar] [CrossRef]
- Hong, G.K.; Kumar, P.; Wang, L.; Damania, B.; Gulley, M.L.; Delecluse, H.J.; Polverini, P.J.; Kenney, S.C. Epstein-Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J. Virol. 2005, 79, 13984–13992. [Google Scholar] [CrossRef] [Green Version]
- Quinlivan, E.B.; Zhang, C.; Stewart, P.W.; Komoltri, C.; Davis, M.G.; Wehbie, R.S. Elevated virus loads of Kaposi’s sarcoma-associated human herpesvirus 8 predict Kaposi’s sarcoma disease progression, but elevated levels of human immunodeficiency virus type 1 do not. J. Infect. Dis. 2002, 185, 1736–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, N.; Duraffour, S.; Snoeck, R.; Andrei, G. KSHV targeted therapy: An update on inhibitors of viral lytic replication. Viruses 2014, 6, 4731–4759. [Google Scholar] [CrossRef] [Green Version]
- Colombini, E.; Guzzo, I.; Morolli, F.; Longo, G.; Russo, C.; Lombardi, A.; Merli, P.; Barzon, L.; Murer, L.; Piga, S.; et al. Viral load of EBV DNAemia is a predictor of EBV-related post-transplant lymphoproliferative disorders in pediatric renal transplant recipients. Pediatr. Nephrol. 2017, 32, 1433–1442. [Google Scholar] [CrossRef]
- Fellner, M.D.; Durand, K.A.; Solernou, V.; Bosaleh, A.; Balbarrey, Z.; Garcia de Davila, M.T.; Rodriguez, M.; Irazu, L.; Alonio, L.V.; Picconi, M.A. Epstein-Barr virus load in transplant patients: Early detection of post-transplant lymphoproliferative disorders. Rev. Argent. Microbiol. 2016, 48, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Malouf, M.A.; Chhajed, P.N.; Hopkins, P.; Plit, M.; Turner, J.; Glanville, A.R. Anti-viral prophylaxis reduces the incidence of lymphoproliferative disease in lung transplant recipients. J. Heart Lung Transplant. 2002, 21, 547–554. [Google Scholar] [CrossRef]
- AlDabbagh, M.A.; Gitman, M.R.; Kumar, D.; Humar, A.; Rotstein, C.; Husain, S. The Role of Antiviral Prophylaxis for the Prevention of Epstein-Barr Virus-Associated Posttransplant Lymphoproliferative Disease in Solid Organ Transplant Recipients: A Systematic Review. Am. J. Transplant. 2017, 17, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Pagano, J.S.; Whitehurst, C.B.; Andrei, G. Antiviral Drugs for EBV. Cancers 2018, 10, 197. [Google Scholar] [CrossRef] [Green Version]
- Andrei, G.; Trompet, E.; Snoeck, R. Novel Therapeutics for Epstein(-)Barr Virus. Molecules 2019, 24, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.R. Epstein-Barr virus (EBV) reactivation and therapeutic inhibitors. J. Clin. Pathol. 2019, 72, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Crumpacker, C.S. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992, 92, 3S–7S. [Google Scholar] [CrossRef]
- Coen, N.; Duraffour, S.; Naesens, L.; Krecmerova, M.; Van den, O.J.; Snoeck, R.; Andrei, G. Evaluation of novel acyclic nucleoside phosphonates against human and animal gammaherpesviruses revealed an altered metabolism of cyclic prodrugs upon Epstein-Barr virus reactivation in P3HR-1 cells. J. Virol. 2013, 87, 12422–12432. [Google Scholar] [CrossRef] [Green Version]
- Coen, N.; Duraffour, S.; Topalis, D.; Snoeck, R.; Andrei, G. Spectrum of activity and mechanisms of resistance of various nucleoside derivatives against gammaherpesviruses. Antimicrob. Agents Chemother. 2014, 58, 7312–7323. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.C.; De Clercq, E.; Pagano, J.S. Inhibitory effects of acyclic nucleoside phosphonate analogs, including (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, on Epstein-Barr virus replication. Antimicrob. Agents Chemother. 1991, 35, 2440–2443. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.K.; Colby, B.M.; Shaw, J.E.; Pagano, J.S. Acyclovir inhibition of Epstein-Barr virus replication. Proc. Natl. Acad. Sci. USA 1980, 77, 5163–5166. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.L. The antiviral prophylaxis of post-transplant lymphoproliferative disorder. Springer Semin. Immunopathol. 1998, 20, 437–453. [Google Scholar] [CrossRef]
- Casper, C.; Wald, A. The use of antiviral drugs in the prevention and treatment of Kaposi sarcoma, multicentric Castleman disease and primary effusion lymphoma. Curr. Top. Microbiol. Immunol. 2007, 312, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Hwang, Y.Y.; Chan, T.S.; Pang, A.W.; Leung, A.Y.; Tse, E.; Kwong, Y.L. Valganciclovir suppressed Epstein Barr virus reactivation during immunosuppression with alemtuzumab. J. Clin. Virol. 2014, 59, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, E.A.; Chillemi, A.C.; Sage, D.R.; Fingeroth, J.D. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 1998, 42, 2923–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deville-Bonne, D.; El Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: Structural and catalytic properties. Antivir. Res. 2010, 86, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Hagemeier, S.R.; Fingeroth, J.D.; Gershburg, E.; Pagano, J.S.; Kenney, S.C. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 2010, 84, 4534–4542. [Google Scholar] [CrossRef] [Green Version]
- Coen, N.; Singh, U.; Vuyyuru, V.; Van den Oord, J.J.; Balzarini, J.; Duraffour, S.; Snoeck, R.; Cheng, Y.C.; Chu, C.K.; Andrei, G. Activity and mechanism of action of HDVD, a novel pyrimidine nucleoside derivative with high levels of selectivity and potency against gammaherpesviruses. J. Virol. 2013, 87, 3839–3851. [Google Scholar] [CrossRef] [Green Version]
- Coen, N.; Duraffour, S.; Haraguchi, K.; Balzarini, J.; van den Oord, J.J.; Snoeck, R.; Andrei, G. Antiherpesvirus activities of two novel 4’-thiothymidine derivatives, KAY-2-41 and KAH-39-149, are dependent on viral and cellular thymidine kinases. Antimicrob. Agents Chemother. 2014, 58, 4328–4340. [Google Scholar] [CrossRef] [Green Version]
- Kenney, S.C. Reactivation and lytic replication of EBV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Fujiwara, S. Animal Models of Human Gammaherpesvirus Infections. Adv. Exp. Med. Biol 2018, 1045, 413–436. [Google Scholar] [CrossRef]
- Rajcani, J.; Kudelova, M. Murine herpesvirus pathogenesis: A model for the analysis of molecular mechanisms of human gamma herpesvirus infections. Acta Microbiol. Immunol. Hung. 2005, 52, 41–71. [Google Scholar] [CrossRef]
- Dong, S.; Forrest, J.C.; Liang, X. Murine Gammaherpesvirus 68: A Small Animal Model for Gammaherpesvirus-Associated Diseases. Adv. Exp. Med. Biol. 2017, 1018, 225–236. [Google Scholar] [CrossRef]
- Martin, M.; Goyette, N.; Boivin, G. Contrasting effects on ganciclovir susceptibility and replicative capacity of two mutations at codon 466 of the human cytomegalovirus UL97 gene. J. Clin. Virol. 2010, 49, 296–298. [Google Scholar] [CrossRef]
- Chevillotte, M.; Schubert, A.; Mertens, T.; von Einem, J. Fluorescence-based assay for phenotypic characterization of human cytomegalovirus polymerase mutations regarding drug susceptibility and viral replicative fitness. Antimicrob. Agents Chemother. 2009, 53, 3752–3761. [Google Scholar] [CrossRef] [Green Version]
- Pesola, J.M.; Coen, D.M. In vivo fitness and virulence of a drug-resistant herpes simplex virus 1 mutant. J Gen. Virol. 2007, 88, 1410–1414. [Google Scholar] [CrossRef]
- Sharma, P.L.; Crumpacker, C.S. Attenuated replication of human immunodeficiency virus type 1 with a didanosine-selected reverse transcriptase mutation. J. Virol. 1997, 71, 8846–8851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Lerma, J.G.; MacInnes, H.; Bennett, D.; Weinstock, H.; Heneine, W. Transmitted human immunodeficiency virus type 1 carrying the D67N or K219Q/E mutation evolves rapidly to zidovudine resistance in vitro and shows a high replicative fitness in the presence of zidovudine. J. Virol. 2004, 78, 7545–7552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.A.; Thompson, M.G.; Paintsil, E.; Ricketts, M.; Gedzior, J.; Alexander, L. Competitive fitness of nevirapine-resistant human immunodeficiency virus type 1 mutants. J. Virol. 2004, 78, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Trompet, E.; Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Viral fitness of MHV-68 viruses harboring drug resistance mutations in the protein kinase or thymidine kinase. Antiviral. Res. 2020, 182, 104901. [Google Scholar] [CrossRef]
- Abram, M.E.; Hluhanich, R.M.; Goodman, D.D.; Andreatta, K.N.; Margot, N.A.; Ye, L.; Niedziela-Majka, A.; Barnes, T.L.; Novikov, N.; Chen, X.; et al. Impact of primary elvitegravir resistance-associated mutations in HIV-1 integrase on drug susceptibility and viral replication fitness. Antimicrob. Agents Chemother. 2013, 57, 2654–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreatta, K.N.; Goodman, D.D.; Miller, M.D.; White, K.L. Reduced viral fitness and lack of cross-class resistance with integrase strand transfer inhibitor and nucleoside reverse transcriptase inhibitor resistance mutations. Antimicrob. Agents Chemother. 2015, 59, 3441–3449. [Google Scholar] [CrossRef] [Green Version]
- Herr, A.J.; Ogawa, M.; Lawrence, N.A.; Williams, L.N.; Eggington, J.M.; Singh, M.; Smith, R.A.; Preston, B.D. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet. 2011, 7, e1002282. [Google Scholar] [CrossRef]
- Herr, A.J.; Williams, L.N.; Preston, B.D. Antimutator variants of DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 548–570. [Google Scholar] [CrossRef] [Green Version]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, F.J.; Knopf, C.W. Herpes simplex virus type 1 DNA polymerase. Mutational analysis of the 3’-5’-exonuclease domain. J. Biol. Chem. 1996, 271, 29245–29254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Garg, P.; Stith, C.M.; Al-Refai, H.; Sterling, J.F.; Murray, L.J.; Kunkel, T.A.; Resnick, M.A.; Burgers, P.M.; Gordenin, D.A. The multiple biological roles of the 3’-->5’ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol. Cell Biol. 2005, 25, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, H.; Scollon, S.; Reuther, J.; Voicu, H.; Rednam, S.P.; Lin, F.Y.; Fisher, K.E.; Chintagumpala, M.; Adesina, A.M.; Parsons, D.W.; et al. Germline POLE mutation in a child with hypermutated medulloblastoma and features of constitutional mismatch repair deficiency. Cold Spring Harb. Mol. Case Stud. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdes-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; Gonzalez, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Shinbrot, E.; Henninger, E.E.; Weinhold, N.; Covington, K.R.; Goksenin, A.Y.; Schultz, N.; Chao, H.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014, 24, 1740–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef]
- Dennis, D.G.; McKay-Fleisch, J.; Eitzen, K.; Dowsett, I.; Kennedy, S.R.; Herr, A.J. Normally lethal amino acid substitutions suppress an ultramutator DNA Polymerase delta variant. Sci. Rep. 2017, 7, 46535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef]
- Leon-Castillo, A.; Britton, H.; McConechy, M.K.; McAlpine, J.N.; Nout, R.; Kommoss, S.; Brucker, S.Y.; Carlson, J.W.; Epstein, E.; Rau, T.T.; et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 2020, 250, 323–335. [Google Scholar] [CrossRef]
- Chen, H.; Beardsley, G.P.; Coen, D.M. Mechanism of ganciclovir-induced chain termination revealed by resistant viral polymerase mutants with reduced exonuclease activity. Proc. Natl. Acad. Sci. USA 2014, 111, 17462–17467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.; Ercolani, R.J.; Lanier, E.R. Novel Cytomegalovirus UL54 DNA Polymerase Gene Mutations Selected In Vitro That Confer Brincidofovir Resistance. Antimicrob. Agents Chemother. 2016, 60, 3845–3848. [Google Scholar] [CrossRef] [Green Version]
- James, S.H.; Prichard, M.N. Current and future therapies for herpes simplex virus infections: Mechanism of action and drug resistance. Curr. Opin. Virol. 2014, 8, 54–61. [Google Scholar] [CrossRef]
- Hakki, M.; Chou, S. The biology of cytomegalovirus drug resistance. Curr. Opin. Infect. Dis. 2011, 24, 605–611. [Google Scholar] [CrossRef] [Green Version]
- Andouard, D.; Mazeron, M.C.; Ligat, G.; Couvreux, A.; Pouteil-Noble, C.; Cahen, R.; Yasdanpanah, Y.; Deering, M.; Viget, N.; Alain, S.; et al. Contrasting effect of new HCMV pUL54 mutations on antiviral drug susceptibility: Benefits and limits of 3D analysis. Antiviral. Res. 2016, 129, 115–119. [Google Scholar] [CrossRef]
- Ducancelle, A.; Champier, G.; Alain, S.; Petit, F.; Le Pors, M.J.; Mazeron, M.C. A novel mutation in the UL54 gene of human cytomegalovirus isolates that confers resistance to foscarnet. Antivir. Ther. 2006, 11, 537–540. [Google Scholar] [PubMed]
- Chou, S. Foscarnet resistance mutations mapping to atypical domains of the cytomegalovirus DNA polymerase gene. Antiviral. Res. 2017, 138, 57–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S. Approach to drug-resistant cytomegalovirus in transplant recipients. Curr. Opin. Infect. Dis. 2015, 28, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.; Devincenzo, J.P.; Toovey, S.; Wu, J.Z.; Whitley, R.J. Comparison of antiviral resistance across acute and chronic viral infections. Antiviral. Res. 2018, 158, 103–112. [Google Scholar] [CrossRef]
- Nijhuis, M.; van Maarseveen, N.M.; Boucher, C.A. Antiviral resistance and impact on viral replication capacity: Evolution of viruses under antiviral pressure occurs in three phases. Handb. Exp. Pharmacol. 2009, 299–320. [Google Scholar] [CrossRef]
- Tu, V.; Abed, Y.; Fage, C.; Baz, M.; Boivin, G. Impact of R152K and R368K neuraminidase catalytic substitutions on in vitro properties and virulence of recombinant A(H1N1)pdm09 viruses. Antiviral. Res. 2018, 154, 110–115. [Google Scholar] [CrossRef]
- Martin, M.; Azzi, A.; Lin, S.X.; Boivin, G. Opposite effect of two cytomegalovirus DNA polymerase mutations on replicative capacity and polymerase activity. Antivir. Ther. 2010, 15, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.W.; Lee, C.P.; Huang, Y.H.; Yang, P.W.; Wang, J.T.; Chen, M.R. Epstein-Barr virus protein kinase BGLF4 targets the nucleus through interaction with nucleoporins. J. Virol. 2012, 86, 8072–8085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.P.; Huang, Y.H.; Lin, S.F.; Chang, Y.; Chang, Y.H.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 2008, 82, 11913–11926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.P.; Chen, J.Y.; Wang, J.T.; Kimura, K.; Takemoto, A.; Lu, C.C.; Chen, M.R. Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. J. Virol. 2007, 81, 5166–5180. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, S.; Murata, T.; Murayama, K.; Yasui, Y.; Sato, Y.; Kudoh, A.; Iwahori, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Epstein-Barr virus polymerase processivity factor enhances BALF2 promoter transcription as a coactivator for the BZLF1 immediate-early protein. J. Biol. Chem. 2009, 284, 21557–21568. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Liao, G.; Shan, L.; Zhang, J.; Chen, M.R.; Hayward, G.S.; Hayward, S.D.; Desai, P.; Zhu, H. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol. 2009, 83, 5219–5231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Pandey, M.; Nandakumar, D.; Raney, K.D.; Yin, Y.W.; Patel, S.S. Excessive excision of correct nucleotides during DNA synthesis explained by replication hurdles. EMBO J. 2020, 39, e103367. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Marousek, G.; Bowlin, T.L. Cyclopropavir susceptibility of cytomegalovirus DNA polymerase mutants selected after antiviral drug exposure. Antimicrob. Agents Chemother. 2012, 56, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.; Marousek, G.I. Accelerated evolution of maribavir resistance in a cytomegalovirus exonuclease domain II mutant. J. Virol. 2008, 82, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, V.; Eckert, K.A. The proofreading 3’–>5’ exonuclease activity of DNA polymerases: A kinetic barrier to translesion DNA synthesis. Mutat. Res. 2002, 510, 45–54. [Google Scholar] [CrossRef]
- Mansky, L.M.; Cunningham, K.S. Virus mutators and antimutators: Roles in evolution, pathogenesis and emergence. Trends Genet. 2000, 16, 512–517. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Bebenek, K. DNA replication fidelity. Annu. Rev. Biochem. 2000, 69, 497–529. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liang, C.; Wu, J.; Liu, L.; Tyo, K.E.J. Increased Processivity, Misincorporation, and Nucleotide Incorporation Efficiency in Sulfolobus solfataricus Dpo4 Thumb Domain Mutants. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Hwang, Y.T.; Lu, Q.; Hwang, C.B. Finger domain mutation affects enzyme activity, DNA replication efficiency, and fidelity of an exonuclease-deficient DNA polymerase of herpes simplex virus type 1. J. Virol. 2009, 83, 7194–7201. [Google Scholar] [CrossRef] [Green Version]
- Andrei, G.; Gammon, D.B.; Fiten, P.; De Clercq, E.; Opdenakker, G.; Snoeck, R.; Evans, D.H. Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice. J. Virol. 2006, 80, 9391–9401. [Google Scholar] [CrossRef] [Green Version]
- Pult, I.; Abbott, N.; Zhang, Y.Y.; Summers, J. Frequency of spontaneous mutations in an avian hepadnavirus infection. J. Virol. 2001, 75, 9623–9632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regoes, R.R.; Hamblin, S.; Tanaka, M.M. Viral mutation rates: Modelling the roles of within-host viral dynamics and the trade-off between replication fidelity and speed. Proc. Biol. Sci. 2013, 280, 20122047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozen-Gagnon, K.; Stapleford, K.A.; Mongelli, V.; Blanc, H.; Failloux, A.B.; Saleh, M.C.; Vignuzzi, M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 2014, 10, e1003877. [Google Scholar] [CrossRef]
- Gnadig, N.F.; Beaucourt, S.; Campagnola, G.; Borderia, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E2294–E2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C. CRISPR/Cas9-Based Antiviral Strategy: Current Status and the Potential Challenge. Molecules 2019, 24, 1349. [Google Scholar] [CrossRef] [Green Version]
- De Silva Feelixge, H.S.; Stone, D.; Roychoudhury, P.; Aubert, M.; Jerome, K.R. CRISPR/Cas9 and Genome Editing for Viral Disease-Is Resistance Futile? ACS Infect. Dis. 2018, 4, 871–880. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Nucleotide Changes | C297W | C981Y | ||||
|---|---|---|---|---|---|---|---|
| Clone A | Clone B | Clone C | Clone A | Clone B | Clone C | ||
| PK 1314 bp | A → C | 1 | |||||
| C → A | 1 | ||||||
| C → T | 1 | ||||||
| G → A | 1 | ||||||
| A → T | 1 | 1 | |||||
| Total nucleotide changes | 0 | 0 | 2 | 1 | 2 | 1 | |
| TK 1935 bp | A → G | 1 | 1 | ||||
| T → C | 1 | 2 | 1 | ||||
| C → A | 1 | 2 | |||||
| C → T | 1 | 1 | |||||
| G → A | 1 | 1 | |||||
| G → T | 1 | ||||||
| A → T | 1 | ||||||
| Frameshift | 1 | ||||||
| Total nucleotide changes | 3 | 5 | 4 | 2 | 1 | 1 | |
| DP 3084 bp | A → C | 1 | |||||
| A → G | 1 | 2 | |||||
| T → C | 2 | 4 | 1 | 1 | |||
| T → G | 1 | 1 | |||||
| C → A | 2 | ||||||
| C → T | 1 | ||||||
| G → A | 1 | ||||||
| G → T | 1 | ||||||
| A → T | 1 | 1 | |||||
| T → A | 1 | 1 | |||||
| Frameshift | 1 | 1 | 1 | ||||
| Total nucleotide changes | 4 | 2 | 14 | 4 | 2 | 0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trompet, E.; Temblador, A.; Gillemot, S.; Topalis, D.; Snoeck, R.; Andrei, G. An MHV-68 Mutator Phenotype Mutant Virus, Confirmed by CRISPR/Cas9-Mediated Gene Editing of the Viral DNA Polymerase Gene, Shows Reduced Viral Fitness. Viruses 2021, 13, 985. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060985
Trompet E, Temblador A, Gillemot S, Topalis D, Snoeck R, Andrei G. An MHV-68 Mutator Phenotype Mutant Virus, Confirmed by CRISPR/Cas9-Mediated Gene Editing of the Viral DNA Polymerase Gene, Shows Reduced Viral Fitness. Viruses. 2021; 13(6):985. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060985
Chicago/Turabian StyleTrompet, Erika, Arturo Temblador, Sarah Gillemot, Dimitrios Topalis, Robert Snoeck, and Graciela Andrei. 2021. "An MHV-68 Mutator Phenotype Mutant Virus, Confirmed by CRISPR/Cas9-Mediated Gene Editing of the Viral DNA Polymerase Gene, Shows Reduced Viral Fitness" Viruses 13, no. 6: 985. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060985