
A Novel Broad Host Range Phage Infecting Alteromonas
by
 , ,
, ,
Xuejin Feng
1, 
Wei Yan
1,2,
Anan Wang
1,
Ruijie Ma
1,
Xiaowei Chen
1,
Ta-Hui Lin
1,
Yi-Lung Chen
1,
Shuzhen Wei
1,
Tao Jin
3,
Nianzhi Jiao
1,* and
Rui Zhang
1,4,* 1
State Key Laboratory of Marine Environmental Science, Fujian Key Laboratory of Marine Carbon Sequestration, College of Ocean and Earth Sciences, Xiamen University, Xiamen 361102, China
2
College of Marine Science and Technology, China University of Geosciences, Wuhan 430074, China
3
Guangzhou Magigene Biotechnology Co., Ltd., Guangzhou 510000, China
4
Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai 519080, China
*
Authors to whom correspondence should be addressed.
Viruses 2021, 13(6), 987; https://0-doi-org.brum.beds.ac.uk/10.3390/v13060987
Submission received: 9 April 2021
/
Revised: 19 May 2021
/
Accepted: 20 May 2021
/
Published: 26 May 2021
(This article belongs to the Special Issue Phage-Host Interactions 2021)
Abstract
:Bacteriophages substantially contribute to bacterial mortality in the ocean and play critical roles in global biogeochemical processes. Alteromonas is a ubiquitous bacterial genus in global tropical and temperate waters, which can cross-protect marine cyanobacteria and thus has important ecological benefits. However, little is known about the biological and ecological features of Alteromonas phages (alterophages). Here, we describe a novel alterophage vB_AmeP-R8W (R8W), which belongs to the Autographiviridae family and infects the deep-clade Alteromonas mediterranea. R8W has an equidistant and icosahedral head (65 ± 1 nm in diameter) and a short tail (12 ± 2 nm in length). The genome size of R8W is 48,825 bp, with a G + C content of 40.55%. R8W possesses three putative auxiliary metabolic genes encoding proteins involved in nucleotide metabolism and DNA binding: thymidylate synthase, nucleoside triphosphate pyrophosphohydrolase, and PhoB. R8W has a rapid lytic cycle with a burst size of 88 plaque-forming units/cell. Notably, R8W has a wide host range, such that it can infect 35 Alteromonas strains; it exhibits a strong specificity for strains isolated from deep waters. R8W has two specific receptor binding proteins and a compatible holin–endolysin system, which contribute to its wide host range. The isolation of R8W will contribute to the understanding of alterophage evolution, as well as the phage–host interactions and ecological importance of alterophages.
1. Introduction
Viruses are widely distributed in the oceans, with an average abundance of approximately 107 viruses mL−1 at the ocean surface [1]. Most marine viruses are bacteriophages that infect bacteria and cause an estimated bacterial mortality rate of 10–50% daily in the ocean [1]. Bacteriophages are exceptionally diverse, both morphologically and genetically; they can facilitate horizontal gene transfer and hence are important for bacterial diversity and evolution [2]. In addition, phage-encoded auxiliary metabolic genes (AMGs), such as fatty acid desaturase genes to regulate the fluidity of host membranes, can enhance phage fitness [3,4,5,6]. Therefore, bacteriophages play crucial roles in marine ecosystems and global biogeochemical cycles [7,8].
The marine bacteria Alteromonas, a genus within the Gammaproteobacteria belonging to a Gram-negative organism, is widely distributed in global tropical and temperate waters [9]. Thus far, 28 species of Alteromonas have been identified. Alteromonas can incorporate the transient nutrients released from phytoplankton to grow rapidly in oligotrophic open oceans [10]. Importantly, although Alteromonas spp. are not always abundant in the environment, they demonstrate high catalase activity and thus can remove hydrogen peroxide from the ocean [11]. Furthermore, Alteromonas can act as important helper bacteria that cross-protect marine cyanobacteria, the most abundant phytoplankton in oligotrophic oceans [12,13]. Hence, Alteromonas spp. are actively involved in the circulation of biogenic matter and the flow of energy in the ocean [12,13].
Despite the ecological importance of the Alteromonas genus, investigations of phages that infect Alteromonas (alterophages) to unveil their genomic features and potential impacts on host bacteria have lagged considerably behind analyses of other marine phages [14,15,16,17,18,19]. For example, more than 100 cyanophages have been published and characterized [20]. Studies of cyanophages and cyanophage–cyanobacteria interactions have greatly advanced the overall understanding of microbial oceanographic processes [4,21]. There are currently 12 published genomes of cultivated marine alterophages, using seven Alteromonas species as hosts (Table 1) [14,15,16,17,18,19]. This limited information regarding alterophages has impeded the overall understanding of the biological and ecological importance of alterophages.
A well-studied model Alteromonas species, Alteromonas macleodii, can be divided into two clades based on physiological and genomic characteristics: surface (A. macleodii) and deep (Alteromonas mediterranea), which are found in ocean surface and deep waters, respectively [22]. In this study, we used the typical deep-clade A. mediterranea as the host to isolate and analyze the novel alterophage vB_AmeP-R8W (R8W). R8W comprises the second known member of the Foturvirus genus within the Autographiviridae family. We carried out a comprehensive survey of host range and performed analyses involving classification and genomic organization of R8W. Importantly, we found that R8W contains various genes that may broaden its host range and affect alterophage–Alteromonas interactions.
2. Materials and Methods
2.1. Phage Isolation, Purification, and Amplification
R8W was isolated from coastal seawater using the double-layer agar method [23]. The water samples were collected from Xiamen Bay (latitude N = 24.253, longitude E = 118.014, depth = 3 m) in October 2014 (Supplementary Materials Figure S1), then filtered through a 0.22 μm membrane (Millipore, Bedford, MA, USA) to remove bacteria and stored in the dark at 4 °C. Host strain A. mediterranea DET was a type strain (DSM17117T = CIP 110805T = LMG 28347T), previously isolated from a depth of 1000 m in the Mediterranean Sea [24]. The host strain was cultured at 28 °C with shaking at 180 rpm in RO medium (200 mL of filtered seawater and 800 mL of artificial seawater supplemented with 0.1% yeast extract, 0.1% peptone, 0.1% sodium acetate, and 0.1% trace metal solution (1 × 10−5 M FeCl3•6H2O, 1 × 10−5 M Na2EDTA•2H2O, 4 × 10−8 M CuSO4•5H2O, 3 × 10−8 M Na2MoO4•2H2O, 8 × 10−8 M ZnSO4•7H2O, 5 × 10−8 M CoCl2•6H2O, and 9 × 10−7 M MnCl2•4H2O)) [23]. Subsequently, 100 mL of host culture was inoculated with the seawater overnight to enrich phages [23]. Thereafter, the phage was filtered and mixed with A. mediterranea DE using the double-layer agar method. After overnight co-culture, clonal plaques were picked from the lawn of host cells and added to 1 mL of SM buffer (50 mM Tris-HCl, 0.1 M NaCl, 8 mM MgSO4, pH 7.5) [23]. These steps were repeated five times to purify the phage. Before DNA extraction, 1 L phage lysate was treated with 2 mg/L of DNase I and RNase A for 1 h at 25 °C, then supplemented with 1 M NaCl for 30 min at 4 °C to promote the separation of phage particles and cell debris. The phage suspension was harvested by centrifugation (10,000× g for 10 min at 4 °C) and passed through 0.22 μm filters, then mixed with 10% polyethylene glycol (PEG8000) and stored for 24 h at 4 °C. The mixture was then centrifuged (10,000× g for 1 h at 4 °C) to precipitate the phage particles. The phage particles were resuspended in 6 mL of SM buffer. High-titer phage suspensions were prepared via CsCl (1.3%, 1.5%, and 1.7%) density gradient centrifugation (200,000× g for 24 h at 4 °C), followed by 30 kDa centrifugal filtration (Millipore, Bedford, MA, USA). The filtrate was stored at 4 °C for subsequent experiments.
2.2. Cross-Infection Experiments
Cross-infections involving phage R8W were performed using 79 Alteromonas strains (see detailed information in Supplementary Materials Table S1). Host cultures (1 mL) undergoing exponential growth were mixed with 5 mL of RO agar medium (0.5% agar, cooled to 45 °C) and poured onto a solid plate of 1.5% RO agar medium. After the agar had solidified, a 1:100 dilution of the phage lysate was aliquoted onto the host lawn (5 μL aliquots) and incubated at 28 °C overnight; SM buffer alone was used as a blank control. Each plaque formation was regarded as successful infection of the corresponding tested strain by R8W.
2.3. One-Step Growth Curve
At a 0.01 multiplicity of infection, the purified phages were adsorbed to 1 mL of exponentially growing host cells in the dark at room temperature for 10 min. Free phages were removed by centrifugation (10,000× g for 5 min at 4 °C). Samples were then resuspended in 100 mL of RO medium for sample collection at 15-min intervals for a total duration of 135 min. Plaques were counted with the double-layer plate method [23]. The co-culture of R8W and host were incubated at 28 °C overnight.
2.4. Transmission Electron Microscopy (TEM)
Phage morphology was analyzed by TEM. Briefly, 20 μL of high-titer phage concentrate was placed on 200-mesh formvar-coated copper electron microscopy grids and allowed to adsorb for 20 min. The phage was negatively stained with 1% (w/v) phosphotungstic acid for 1 min; the excess stain was removed with filter paper and then air dried for 2 h. The phage was imaged using a Tecnai G2 Spirit BioTwin TEM (FEI Thermo Fisher Scientific, Eindhoven, The Netherlands) at 120 kV.
2.5. Genome Sequencing, Assembly and Annotation
The phage was lysed by incubation with 50 μM proteinase K, 20 mM EDTA, and 0.5% SDS at 65 °C for 3 h. The phage DNA was extracted using the phenol-chloroform method. DNA was sonicated using a Covaris to an average length of 350 bp. DNA fragments were then end repaired, 3′-adenylated, and amplified using Illumina sequencing adapter-specific primers. After quality control, quantification and normalization of the DNA libraries, 150 bp paired-end reads were generated from the Illumina Novaseq. Raw reads were trimmed using Trimmomatic version 0.36 [25] (parameters: version 0.36, ILLUMINACLIP: TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:40) to gain clean reads, which comprised more than 90% of the raw data. Bowtie2 version 2.3.4 [26] was used to remove sequences attributed to the host bacterial genome; high-quality clean reads were then assembled using IDBA-UD version 1.1.3 (parameters: kmer min 21 max 91 step 10) [27]. The termini were identified by PhageTerm [23,28]. The reads with the maximum coverage were considered as phage termini, which was “5′-CTGGGCACTAACCCACACAACGTACCATAT-3′”. The complete genome sequence was submitted to the GenBank database under accession number MW043865.
The genes in assembled genomic sequences were predicted by PRODIGAL [29], GeneMarkS [30] (http://topaz.gatech.edu/GeneMark/genemarks.cgi, accessed on 9 April 2021), and RAST (http://rast.nmpdr.org/, accessed on 9 April 2021) [31]. Annotation of the functions of the predicted gene products were conducted using BLAST search algorithms (parameters: e-value <0.001). The tRNAscan-SE program was used to predict tRNA sequences [32].
The receptor binding proteins (RBPs) were identified using a previously described method [33]. The functions of the predicted R8W gene products that were annotated as tail fibers, tail spikes, and hypothetical proteins were analyzed using Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index, accessed on 9 April 2021) (parameters: confidence > 40%) [34], UniProtKB (https://www.uniprot.org, accessed on 9 April 2021) (parameters: e-value < 0.01), Pfam (http://pfam.xfam.org/search/sequence, accessed on 9 April 2021) (parameters: e-value < 0.01) [35], and HHPred (https://toolkit.tuebingen.mpg.de, accessed on 9 April 2021) (parameters: e-value < 0.001, cols > 80) to predict depolymerase activity [36]. The structures were predicted using the I-TASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/, accessed on 9 April 2021) and visualized using PyMOL [37,38].
2.6. Phylogenetic Analysis
The large terminase subunit (TerL) and thymidylate synthase (ThyX and ThyA) proteins were used for phylogenetic analysis. The TerL protein is conserved in Caudovirales [39]. ThyX and ThyA are encoded by putative AMG and are present in many viral and bacterial genomes [40]. Phylogenetic analyses of these two proteins were used to assess the genetic distances of members within the Autographiviridae. Individual amino acid sequences of the proteins were aligned using Mafft version 7.313 [41] (parameters: –adjustdirectionaccurately –auto), and a maximum-likelihood phylogenetic tree was constructed using RAxML version 8.2.11 with a bootstrap value of 1000 (parameters: -f a -m PROTGAMMAWAG -N 1000) [42]. The average nucleotide identity (ANI) was calculated using OrthANI software [43] and JSpeciesWS (http://jspecies.ribohost.com/jspeciesws/#analyse, accessed on 9 April 2021) [44] to establish ANI phylogenetic trees.
The classification of R8W among 57 reference genomes (i.e., most currently known Autographiviridae viruses, 12 alterophages (see detailed information in Table 1), and other similar viruses) was performed using the Virus Classification and Tree Building Online Resource (VICTOR, https://victor.dsmz.de, accessed on 9 April 2021) as described by Meier-Kolthoff and colleagues [45,46]. Briefly, all pairwise comparisons of viral nucleotide sequences were conducted using the genome BLAST distance phylogeny method under settings recommended for prokaryotic viruses [46,47]. The taxonomies at levels of species, genus, subfamily, and family were estimated using the OPTSIL program with the recommended clustering thresholds and an F-value (fraction of links required for cluster fusion) of 0.5 [45,46].
3. Results and Discussion
3.1. Biological Characteristics
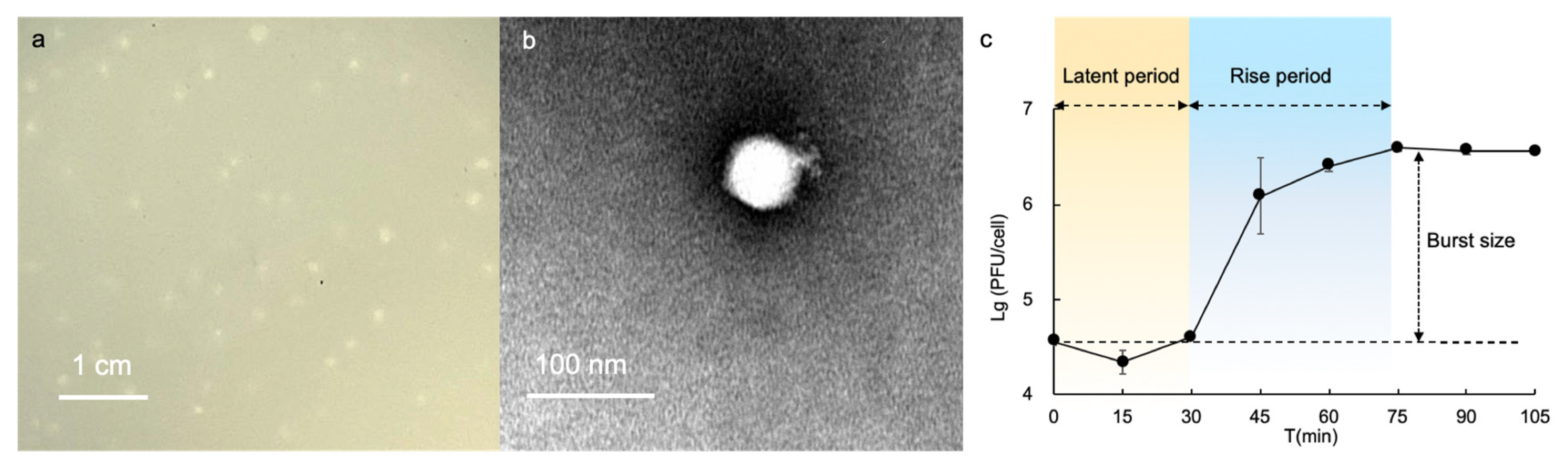
A novel alterophage R8W was isolated from Xiamen Bay, China, using the deep-clade A. mediterranea DE as a host. After 24 h of infection with purified R8W, slightly round plaques with diameters <1 mm were produced, indicating the presence of virions released from lysed cells (Figure 1a). Additionally, TEM was used to characterize the morphology of R8W; these analyses revealed that R8W has an equidistant and icosahedral head (65 ± 1 nm in diameter) with a short tail (12 ± 2 nm in length) (Figure 1b).
We determined the lytic cycle of R8W with a one-step growth curve at 0.01 multiplicity of infection. This growth curve showed a latent period of approximately 30 min, followed by a rise period of 45 min and a burst size of 88 plaque-forming units/cell (Figure 1c). These findings indicated that the burst size and rise period of R8W differed from those of previously described alterophages [15,16,19]. For example, alterophages AltAD45-P1 and P2 have a rise period of approximately 5 h [19].
The ability of R8W to cross-infect other Alteromonas spp. was investigated. The tested strains included a mostly complete collection of known Alteromonas type strains and a considerable number of A. macleodii isolates. We used various phage abundances (105, 107, 109, and 1011 plaque-forming units/mL) to determine the efficiency of infection. To the best of our knowledge, R8W exhibits the broadest host range among all known alterophages. Previous studies have shown that alterophages generally exhibit a narrow host range. For example, two siphophages (JH01 and PB15) and one myophage (vB_AmeM_PT11-V22) only infected their original hosts [14,16,18]. Furthermore, four N4-like podophages (vB_AmaP_AltAD45-P1, P2, P3, and P4) were able to infect two Alteromonas, A. mediterranea and A. macleodii [19]. In our study, R8W could infect 35 of the 79 strains tested (infection rate of 44.30%); these 35 strains included nine species (Table 2).
According to their isolation sources, the Alteromonas spp. in this study were divided into five categories, including surface seawaters, subsurface seawaters, deep seawaters, sediment, and other origins. The respective infection rates were 46%, 44%, 50%, 36%, and 37.5%. Therefore, R8W exhibited particularly strong specificity for strains isolated from deep seawaters (Table 2). Additionally, the 35 tested strains that could be infected by R8W had diverse global geographical origins (Table 2). The rate of R8W infection among Alteromonas spp. isolated from surface and subsurface seawaters was nearly 50%. Overall, these results suggest that R8W can survive in multiple oceanic regions and might be widely distributed in the global ocean.
3.2. Genomic Characteristics
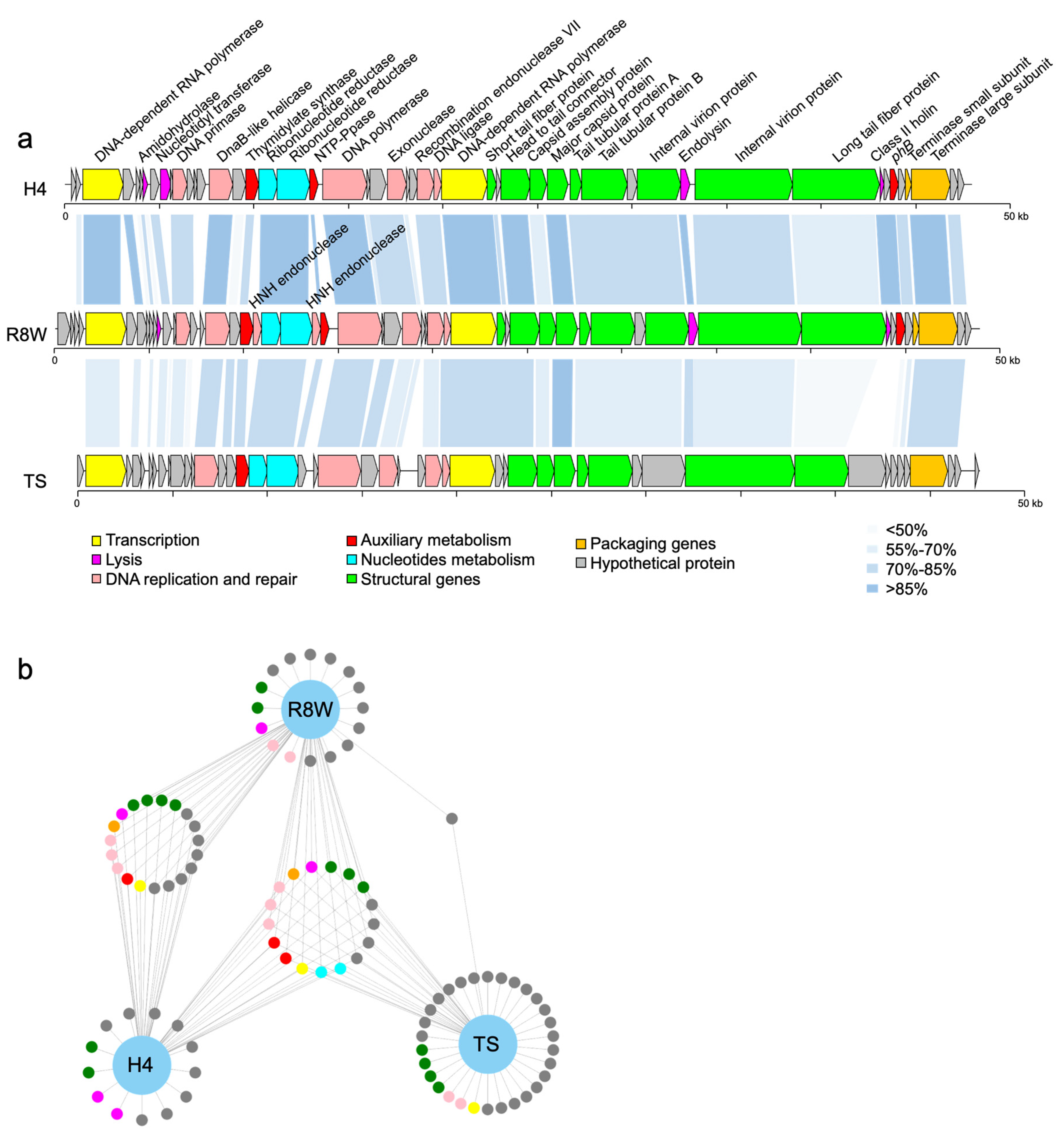
Analysis of the R8W genome revealed 48,825 bp of double-stranded DNA with a G+C content of 40.55%. R8W has a linear genome with direct terminal repeats of 202 bp (Supplementary Data 1). R8W belongs to the T7-like phages, in which the direct terminal repeats are recognized by the terminase as a fixed site and generated by DNA replication during packaging [28]. The R8W genome has 53 putative protein-coding sequences, which comprise 96.95% of the genome, with amino acid sequence lengths of 35 to 1818. Of those putative protein-coding sequences, 24 gene products were identified as hypothetical proteins, while 29 gene products were arranged in eight functional categories (Figure 2a and Supplementary Materials Table S2), including auxiliary metabolism, DNA interactions, signal transduction regulation, and packaging. Among the genomic loci, predicted DNA replication and repair proteins occupied the largest proportion (31.24%). No tRNA sequences were identified in the R8W genome using the tRNAscan-SE program, suggesting that there may be close interactions between the phage and its host during protein synthesis [48].
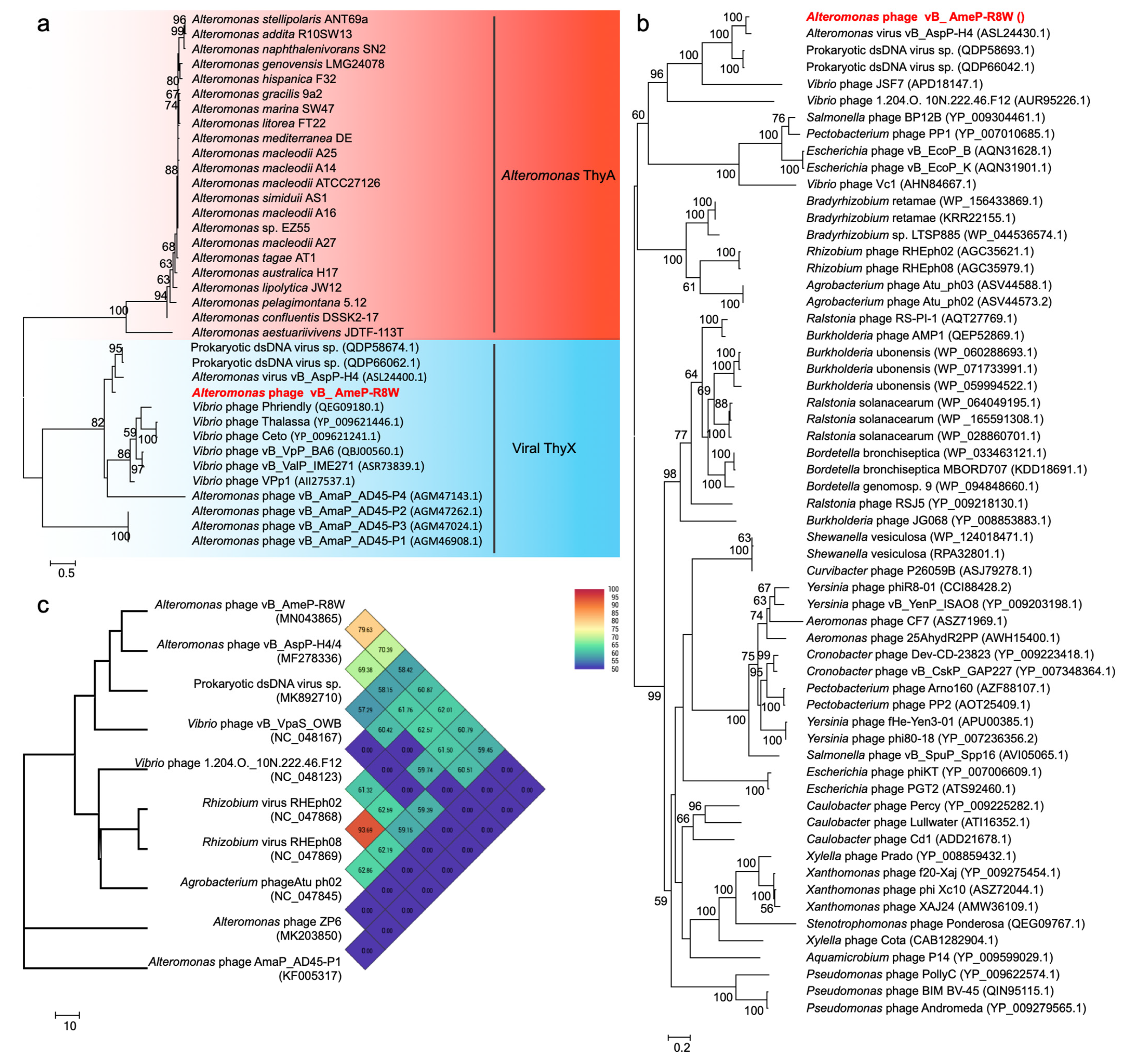
Phage-encoded AMGs can directly modulate host function and enhance viral propagation, subsequently altering biogeochemical cycling. AMGs have been widely reported in cyanophages, where they are involved in photosynthesis and nitrogen metabolism [3,4,5]. In this study, we identified three putative AMGs in the R8W genome: g19, g24, and g48. g19 encodes ThyX, a thymidylate synthase with two forms, ThyX and ThyA, which are primarily found in phages and bacteria, respectively. Thymidylate synthase can catalyze the synthesis of thymidylate and bind RNA to inactivate regulatory gene expression [49,50]. We built a phylogenetic tree of thymidylate synthase based on amino acid sequences in which the known proteins were divided into two phylogenetic groups: viral ThyX and Alteromonas ThyA (Figure 3a). g24 encodes a nucleoside triphosphate pyrophosphohydrolase, which exhibits robust similarity to dUTP pyrophosphatase and nucleoside triphosphate pyrophosphohydrolase MazG proteins; however, it only consists of a single MazG-like domain, which may influence the host signal and survival rate during phage reproduction [51]. These two genes were previously found in other phages, including Vibrio phages, Roseobacter phages, and cyanophages [40,52]. G48 is annotated as the putative DNA-binding response regulator, PhoB, which functions as a two-component response regulator [53,54]. Phosphorylation of PhoB can enhance the affinity of DNA binding to regulate transcription in low-phosphate environments [53,54]. The presence of phoB gene in the R8W genome implies potential adaptations to low-phosphate environments. These three AMGs were associated with viral nucleotide metabolism and host metabolism, potentially improving DNA replication efficiency by enabling R8W to utilize its own genes to acquire the metabolites needed during phage infection.
Comparative genomic analysis revealed that R8W has isomorphic modules and gene synteny similar to those of alterophage vB_AspP-H4/4 (H4) and a prokaryotic virus in a metagenome-assembled bin (TS) (Figure 2a). Phage H4 (GenBank accession no. MF278336) was isolated from North Sea water using Alteromonas addita as its host [17]. TS (GenBank accession no. MK892710) was assembled from viral metagenome sequences from the Tara Oceans expedition at station TARA_022 (latitude N = 39.8386, longitude E = 17.4155, depth = 5 m) [5]. Genome maps showed that structural proteins are concentrated in the middle and right-hand regions of R8W, while genes for DNA replication and repair are located upstream of the structural genes. Furthermore, nucleotide metabolism genes and AMGs are distributed among the DNA replication and repair genes. Based on the total amino acid matches, R8W has remarkable similarity with H4 and TS (29.56–98.55% and 38.67–85.99% identities, respectively). In particular, nucleotide metabolism genes, packaging genes, and AMGs were common among the three phages (identity > 70%) (Figure 2b, Supplementary Materials Table S3); these gene products are important for ensuring full phage assembly and host interactions [23,40,55].
3.3. Broad Host Range of Phage R8W
Adsorption of phages to their hosts is the first step in the lytic cycle [56]. Phages have been found to encode RBPs, which are a critical structure in host-specific recognition [14,56]. RBPs are usually located at the distal end of the tail along with tail fiber proteins, tail spike proteins, and tail tip proteins [14,56]. Including R8W, all the 11 known alterophages can infect 10 different Alteromonas species [14,15,16,17,18,19]. Our analysis demonstrated 13 types of RBPs in these alterophages (Table 3), including pectin degradation protein, glycoside hydrolase, endo-N-acetyl neuraminidase, glucosidase, xylosidase, and glucanase. They may specifically recognize host flagellum glycosylation, capsular exopolysaccharide, and lipopolysaccharides [22]. These genes were potential phages receptors [22]. In a previous study, Klebsiella phages were found to exhibit 11 types of RBPs with depolymerase activity, which enabled infection of distinct Klebsiella pneumoniae strains with thick polysaccharide capsules [33]. The Escherichia coli phage phi92 contains five types of RBPs, two of which have degrading enzyme activity (endo-N-acyl neuraminidase and colanidase). This phage can infect eight E. coli strains and 19 Salmonella strains, thus demonstrating a broad host range [57].
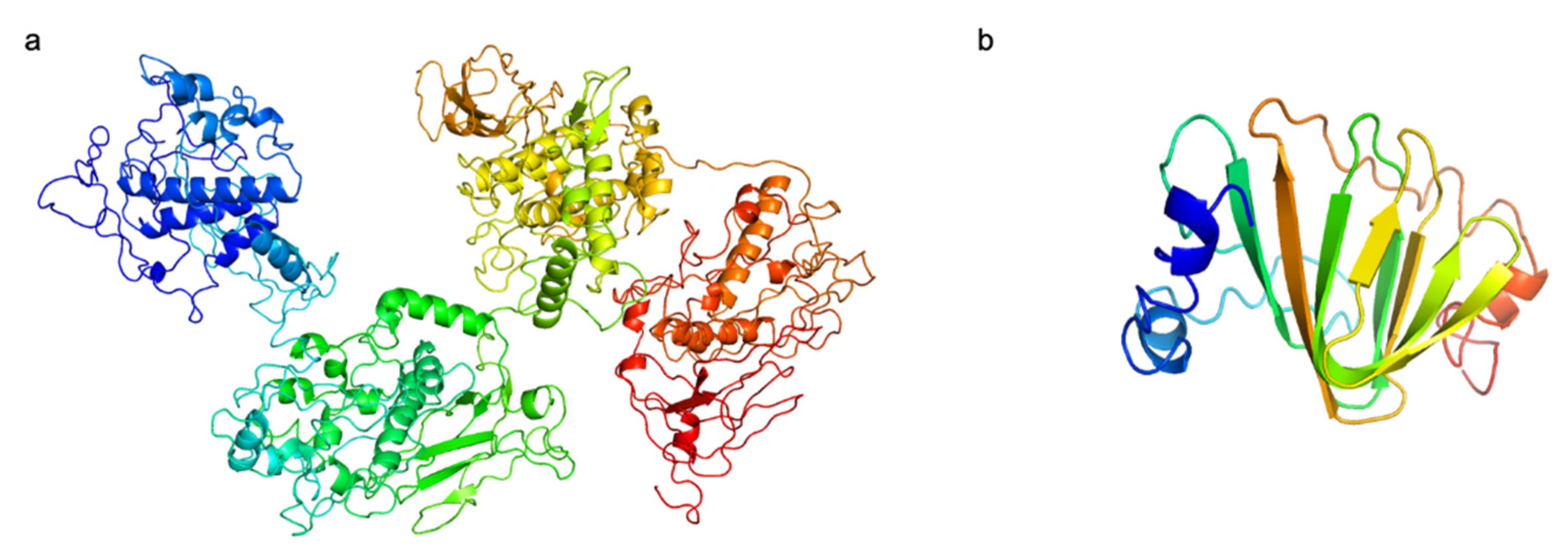
Two putative RBPs were identified in the R8W genome, including Gp34 and Gp45; their predicted structures are shown in Figure 4. First, their functions were annotated as putative tail fibers. The N-terminal domain of Gp45 has a high degree of identity to the T7-tail fiber protein, as indicated by Pfam (bit score, 30.5; e-value, 2.5 × 10−7). T7-tail fiber proteins recognize bacterial outer membrane lipopolysaccharide [58,59]. Second, the C-terminal domain of Gp45 was predicted to be a glycoside hydrolase by HHpred (probability, 96.68%; e-value, 0.062; aligned cols, 171) and Phyre2 (confidence, 91.3%; identity, 27%); the C-terminal domain of Gp34 was predicted to be a dimethylsulfoniopropionate lyase by HHpred (probability, 99.11; e-value, 5.3 × 10−9; aligned cols, 94) and Phyer2 (confidence, 92.8%; identity, 18%). These results suggest that Gp34 and Gp45 may have depolymerization activity that enables hydrolysis of bacterial exopolysaccharide, which may be the first step in the R8W infection process. Indeed, Alteromonas gracilis 9a2, an exopolysaccharide producer, is efficiently infected by R8W. The exopolysaccharide of A. gracilis 9a2 consists of mannose, galactose, and glucose, which may be one of the R8W receptors, and be recognized by glycosyl hydrolase in R8W, thus facilitating R8W infection [60]. Thus, we speculate that its possession of two types of RBPs with depolymerization activity might contribute to the broader host range of R8W. The RBPs of the alterophages JH01 and vB_AmeM_PT11-V22 are capsid fiber protein and tail fiber protein, respectively. Additionally, the alterophages JH01 and vB_AmeM_PT11-V22 are only able to infect their original hosts, A. marina SW-47 and A. mediterranea PT11, respectively. [14,18]. Alterophage PB15 contains two RBPs and may infect a wide range of hosts, although the original study showed a narrow host range among the few host strains tested [16].
The host range of R8W may also be determined by its lytic ability to exit the host [56]. In the R8W genome, g43 and g46 are predicted to be putative endolysin and class II holin genes, respectively. These two gene products enable phages to initiate lysis at a specific point during the infection [56,61]. Holin protein can insert into the host cytoplasmic membrane, oligomerize, and form holes in the membrane [56,61]. Then, the endolysin passes through these holes to selectively degrade peptidoglycan [56,61]. The presence or absence of a holin gene in Lactococcus lactis phages affects their lytic efficiency [62]. The holin–endolysin system has also been found in alterophages vB_AcoS-R7M, ZP6 and H4. Thus, we infer that vB_AcoS-R7M, ZP6 and H4 have broad host ranges.
3.4. R8W Is Characterized as a New Species of Foturvirus Genus within the Autographiviridae Family
To evaluate the genetic relationships of R8W, we constructed a phylogenetic tree using 60 similar amino acid sequences of TerL (Figure 3b). R8W and H4 were grouped together into a clade containing two prokaryotic viruses in metagenome-assembled bins (TS and TS1) and two Vibrio phages, suggesting that these phages are close relatives. TS1 (GenBank accession no. MK892670) was assembled from viral metagenome sequences collected during the Tara Oceans expedition at station TARA_052 (latitude N = −16.957, longitude E = 53.9801). R8W and its closest relative H4 exhibited distinct genomic information and lower ANI values (69.38–79.63%) (Figure 3c); these findings implied that R8W is a novel alterophage species. BLASTN analysis also showed that the R8W genome was closely related to the H4 and TS genomes with sequence identities of 79.16% and 78.64%, respectively.
Phages are regarded as members of the same genus by the International Committee on Taxonomy of Viruses when their nucleotide sequence identities are greater than 50% [63]. The ANI values were between 69.38% and 79.63% for comparisons of R8W, H4, and TS (Figure 3c), suggesting that these phages can be grouped into a single genus but should be regarded as different species [40,63]. However, the application of ANI to taxonomy is limited to sequences with high coverage at the genome-wide level. We selected the 45 genomes of Autographiviridae viruses, 12 alterophages, and TS to reconstruct the genome BLAST distance phylogeny tree (Figure 5a). OPTSIL clustering produced 48 species clusters, 17 genera clusters, and 8 family clusters (Figure 5a,b). The tree showed reliable bootstrap values at most nodes. Both genome sizes and G + C contents of distinct species were strongly correlated by means of phylogenetic clustering (Figure 5a,c). The results of phylogenetic clustering were also consistent with the classifications defined by the International Committee on Taxonomy of Viruses (Figure 5a,b). The phylogenetic trees implied that R8W is closely related to Vibrio and Rhizobium phages; this relationship is also supported by phylogenetic analysis of the core protein, TerL (Figure 3c). The above results indicate that R8W is appropriately classified as a new species; moreover, R8W, H4, and TS belong to the Foturvirus genus in the Autographiviridae family.
4. Conclusions
In this study, we isolated and defined the novel alterophage species vB_AmeP-R8W using a deep-clade Alteromonas mediterranea as its host. This novel alterophage species belongs to the Autographiviridae family and thus extends our knowledge of alterophage–host interactions. This large survey concerning alterophage host range showed that R8W has a broad host range and has particularly strong specificity for Alteromonas strains isolated from deep waters. Numerous important functional proteins were found in R8W, such as RBPs, which potentially increase its replication success and host recognition. R8W possesses specific RBPs and a compatible holin–endolysin system to expand its host range. However, there were some limitations in terms of the proteomic analysis and structural resolution of R8W, which limited the ability to verify alterophage–Alteromonas interactions. Considering the importance of Alteromonas and the influences of alterophages on their hosts, more alterophages must be isolated to analyze their genomic and evolutionary diversities. In the future, more alterophage–Alteromonas model systems could be investigated, which will greatly enhance the overall knowledge regarding the ecological implications of alterophages and their hosts.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/v13060987/s1, Table S1. Genomic information of Alteromonas strains used in this study; Table S2. vB_AmeP-R8W (R8W) gemone annotation; Table S3. The genetic comparison of the phage vB_AmeP-R8W (R8W), vB_AspP-H4/4 (H4) and prokaryotic dsDNA virus (TS) (similarity > 70%); Figure S1. Station map indicating location where vB_AmeP-R8W (R8W) was isolated. Supplementary Data 1. Results of R8W PhageTerm analysis.
Author Contributions
The sponsors had no role in the design, execution, interpretation, or writing of the study. R.Z. and N.J. designed the study. X.F. conducted the culture and biological characteristics of R8W. A.W. conducted the isolation of R8W. X.F., T.J. and S.W. participated in genome analysis. X.F., W.Y., R.M., X.C., T.-H.L., Y.-L.C., R.Z. and N.J. wrote the manuscript. All authors contributed to the final version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (91951209, 41861144018, 41906110) and the National Key Research and Development Program of China (2020YFA06083000). R.Z. was partially supported by the Fundamental Research Funds for the Central Universities (20720200027) and the Science and Technology Program of Guangzhou, China (201904020029).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The complete genome sequence of alterophage vB_AmeP-R8W was submitted to the GenBank database under accession number MW043865.
Acknowledgments
R.M. and X.C. were supported by The PhD Fellowship of the State Key Laboratory of Marine Environmental Science at Xiamen University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol 2015, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.H.; Cook, A.; Millard, A.; Bailey, S.; Clokie, M. Bacterial photosynthesis genes in a virus. Nature 2003, 424, 741. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Roitman, S.; Hornung, E.; Flores-Uribe, J.; Sharon, I.; Feussner, I.; Béjà, O. Cyanophage-encoded lipid desaturases: Oceanic distribution, diversity and function. Int. Soc. Microb. Ecol. J. 2018, 12, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, A.E.; Howard-Varona, C.; Needham, D.M.; John, S.G.; Worden, A.Z.; Sullivan, M.B.; Waldbauer, J.R.; Coleman, M.L. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat. Rev. Microbiol. 2020, 18, 21–34. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- López-Pérez, M.; Gonzaga, A.; Martin-Cuadrado, A.B.; Onyshchenko, O.; Ghavidel, A.; Ghai, R.; Rodriguez-Valera, F. Genomes of Surface Isolates of Alteromonas macleodii: The life of a widespread marine opportunistic copiotroph. Sci. Rep. 2012, 2, 696. [Google Scholar] [CrossRef] [Green Version]
- Neumann, A.M.; Balmonte, J.P.; Berger, M.; Giebel, H.; Arnosti, C.; Voget, S.; Simon, M.; Brinkhoff, T.; Wietz, M. Polysaccharide utilization by A. macleodii. Environ. Microbiol. 2015, 17, 3857–3868. [Google Scholar] [CrossRef] [PubMed]
- Zinser, E.R. The microbial contribution to reactive oxygen species dynamics in marine ecosystems. Env. Microbiol. Rep. 2018, 10, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.J.; Kirkegaard, R.; Szul, M.J.; Johnson, Z.I.; Zinser, E.R. Facilitation of robust growth of Prochlorococcus colonies and dilute liquid cultures by “helper” heterotrophic bacteria. Appl. Environ. Microbiol. 2008, 74, 4530–4534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinser, E.R. Cross-protection from hydrogen peroxide by helper microbes: The impacts on the cyanobacterium Prochlorococcus and other beneficiaries in marine communities. Env. Microbiol. Rep. 2018, 10, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Serrano, R.; Dunne, M.; Rosselli, R.; Martin-Cuadrado, A.-B.; Grosboillot, V.; Zinsli, L.V.; Roda-Garcia, J.J.; Loessner, M.J.; Rodriguez-Valera, F. Alteromonas Myovirus V22 represents a new genus of marine bacteriophages requiring a tail fiber chaperone for host recognition. Msystems 2020, 5, e00217-20. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Wang, M.; Wang, M.; Jiang, T.; Sun, J.; Gao, C.; Jiang, Y.; Guo, C.; Shao, H.; et al. Characterization and genome analysis of a novel marine Alteromonas phage P24. Curr. Microbiol. 2020, 77, 2813–2820. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Q.; Wang, M.; Zhao, G.; Jiang, Y.; Malin, G.; Gong, Z.; Meng, X.; Liu, Z.; Lin, T.; et al. Characterization and genome sequence of marine Alteromonas gracilis phage PB15 isolated from the Yellow Sea, China. Curr. Microbiol. 2017, 74, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Kallies, R.; Kiesel, B.; Zopfi, J.; Wick, L.Y.; Chatzinotas, A. Complete genome sequence of Alteromonas virus VB_AspP-H4/4. Genome Announc. 2017, 5, e00914-17. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Jiang, Y.; Xiao, S.; Wang, M.; Liu, Q.; Huang, L.; Xue, C.; Wang, Q.; Lin, T.; Shao, H.; et al. Characterization and genome analysis of a novel Alteromonas phage JH01 isolated from the Qingdao coast of China. Curr. Microbiol. 2019, 76, 1256–1263. [Google Scholar] [CrossRef]
- Garcia-Heredia, I.; Rodriguez-Valera, F.; Martin-Cuadrado, A. Novel group of podovirus infecting the marine bacterium Alteromonas macleodii. Bacteriophage 2014, 3, e24766. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Jin, H.; Wang, X.; Li, Q.; Zhang, J.; Cui, N.; Jiang, Y.; Chen, Y.; Wu, Q.; Zhou, C.; et al. Genomic analysis of mic1 reveals a novel freshwater long-tailed cyanophage. Front. Microbiol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Laurenceau, R.; Raho, N.; Forget, M.; Arellano, A.A.; Chisholm, S.W. Frequency of mispackaging of Prochlorococcus DNA by cyanophage. Int. Soc. Microb. Ecol. J. 2021, 15, 129–140. [Google Scholar] [CrossRef]
- López-Pérez, M.; Rodriguez-Valera, F. Pangenome evolution in the marine bacterium Alteromonas. Genome Biol. Evol. 2016, 8, 1556–1570. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Ma, R.; Chen, H.; Yang, Y.; Jiao, N.; Zhang, R. A newly isolated roseophage represents a distinct member of Siphoviridae family. Virol. J. 2019, 16, 128. [Google Scholar] [CrossRef]
- Ivanova, E.P.; López-Pérez, M.; Zabalos, M.; Nguyen, S.H.; Webb, H.K.; Ryan, J.; Lagutin, K.; Vyssotski, M.; Crawford, R.J.; Rodriguez-Valera, F. Ecophysiological diversity of a novel member of the genus Alteromonas, and description of Alteromonas mediterranea sp. nov. Anton. Leeuw. Int. J. 2015, 107, 119–132. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-se on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the architecture of depolymerase-containing receptor binding proteins in Klebsiella phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, gky995. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely reimplemented MPI bioinformatics toolkit with a new HHpred Server at its core. J. Mol. Biol. 2017, 430, 2237–2243. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER Server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, L.; Yu, D.; Du, S.; Zhang, L.; Zhang, L.; Wu, C.; Xiong, C.; Han, L.; He, J. Diversity and potential biogeochemical impacts of viruses in bulk and rhizosphere soils. Environ. Microbiol. 2021, 23, 588–599. [Google Scholar] [CrossRef]
- Nilsson, E.; Li, K.; Fridlund, J.; Šulčius, S.; Bunse, C.; Karlsson, C.M.G.; Lindh, M.; Lundin, D.; Pinhassi, J.; Holmfeldt, K. Genomic and seasonal variations among aquatic phages infecting the Baltic Sea gammaproteobacterium Rheinheimera sp. strain BAL341. Appl. Environ. Microbiol. 2019, 85, e01003-19. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, Y.O.; Park, S.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia Coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Delesalle, V.A.; Tanke, N.T.; Vill, A.C.; Krukonis, G.P. Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 2016, 6, e1219441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asare, P.T.; Jeong, T.; Ryu, S.; Klumpp, J.; Loessner, M.J.; Merrill, B.D.; Kim, K. Putative type 1 thymidylate synthase and dihydrofolate reductase as signature genes of a novel bastille-like group of phages in the subfamily Spounavirinae. BMC Genom. 2015, 16, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Suh, S.W.; Song, H.K. A cytosine modification mechanism revealed by the structure of a ternary complex of deoxycytidylate hydroxymethylase from bacteriophage T4 with its cofactor and substrate. IUCrJ 2019, 6, 206–217. [Google Scholar] [CrossRef]
- Bryan, M.J.; Burroughs, N.J.; Spence, E.M.; Clokie, M.R.J.; Mann, N.H.; Bryan, S.J. Evidence for the intense exchange of mazG in marine cyanophages by horizontal gene transfer. PLoS ONE 2008, 3, e2048. [Google Scholar] [CrossRef] [Green Version]
- Katharios, P.; Kalatzis, P.G.; Kokkari, C.; Sarropoulou, E.; Middelboe, M. Isolation and characterization of a N4-like lytic bacteriophage infecting Vibrio splendidus, a pathogen of fish and bivalves. PLoS ONE 2017, 12, e0190083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Chisholm, S.W. Marine viruses exploit their host’s two-component regulatory system in response to resource limitation. Curr. Biol. 2012, 22, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Kou, X.; Liu, X.; Liu, Y.; Li, C.; Liu, M.; Jiang, L. Backbone resonance assignment of the response regulator protein PhoBNF20D from Escherichia coli. Biomol. NMR Assigm. 2018, 12, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Huang, S.; Voget, S.; Simon, M.; Chen, F. A Novel Roseobacter phage possesses features of Podoviruses, Siphoviruses, prophages and gene transfer agents. Sci. Rep. 2016, 6, 30372. [Google Scholar] [CrossRef]
- de Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and evolutionary determinants of bacteriophage host range. Trends Microbiol. 2018, 27, 51–63. [Google Scholar] [CrossRef]
- Schwarzer, D.; Buettner, F.F.R.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.; Bowman, V.D.; Stummeyer, K.; Mühlenhoff, M.; et al. A multivalent adsorption apparatus explains the broad host range of phage phi92: A comprehensive genomic and structural analysis. J. Virol. 2012, 86, 10384–10398. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Doval, C.; Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzman, T.; Globus, R.; Molshanski-Mor, S.; Ben-Shem, A.; Yosef, I.; Qimron, U. A continuous evolution system for contracting the host range of bacteriophage T7. Sci. Rep. 2020, 10, 307. [Google Scholar] [CrossRef]
- Matsuyama, H.; Minami, H.; Sakaki, T.; Kasahara, H.; Baba, S.; Ishimaru, S.; Hirota, K.; Yumoto, I. Alteromonas gracilis sp. nov., a marine polysaccharide-producing bacterium. Int. J. Syst. Evol. Micr. 2015, 65, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Loessner, M.J. Bacteriophage endolysins-current state of research and applications. Curr. Opin. Microbiol. 2005, 8, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.; Vukov, N.; Loessner, M.J.; Moineau, S. Distribution and composition of the lysis cassette of Lactococcus lactis phages and functional analysis of bacteriophage ul36 holin. FEMS Microbiol. Lett. 2004, 233, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Biological characteristics of the alterophage vB_AmeP-R8W (R8W). (a) Image of plaques 24 h after infection. (b) TEM of the podovirus alterophage R8W. (c) One-step growth curve of R8W in the host, Alteromonas mediterranea strain DE. Error bars indicate standard deviation.
Figure 1.
Biological characteristics of the alterophage vB_AmeP-R8W (R8W). (a) Image of plaques 24 h after infection. (b) TEM of the podovirus alterophage R8W. (c) One-step growth curve of R8W in the host, Alteromonas mediterranea strain DE. Error bars indicate standard deviation.

Figure 2.
(a) Genome organization and comparison of the phage R8W to vB_AspP-H4/4 (H4) (GenBank accession no. MF278336) and prokaryotic dsDNA virus TS (GenBank accession no. MK892710, isolated from the Tara Oceans expedition Tp1_25_SUR_0-0d2_C3569776_1). Arrows indicate the direction of transcription of each gene. Each color indicates a putative function. The color gradients represent the amino acid sequence identity obtained from BLASTP matching. (b) The network diagram shows the similarity between three phage genomes. The nodes represent genes, and the other end is a phage connected by lines. Genes from different phages that are in the same loop indicate that the similarity between the two genes is greater than 70%.
Figure 2.
(a) Genome organization and comparison of the phage R8W to vB_AspP-H4/4 (H4) (GenBank accession no. MF278336) and prokaryotic dsDNA virus TS (GenBank accession no. MK892710, isolated from the Tara Oceans expedition Tp1_25_SUR_0-0d2_C3569776_1). Arrows indicate the direction of transcription of each gene. Each color indicates a putative function. The color gradients represent the amino acid sequence identity obtained from BLASTP matching. (b) The network diagram shows the similarity between three phage genomes. The nodes represent genes, and the other end is a phage connected by lines. Genes from different phages that are in the same loop indicate that the similarity between the two genes is greater than 70%.

Figure 3.
(a) Phylogenetic analysis of thymidylate synthase amino acid sequences of 22 Alteromonas and 14 phages. (b) Phylogenetic analysis of TerL amino acid sequences of 60 phages. (c) The genome-wide tree based on the average nucleotide identity (ANI) from 10 phages. Numbers at the nodes indicate bootstrap values (1000 replications and values >50%).
Figure 3.
(a) Phylogenetic analysis of thymidylate synthase amino acid sequences of 22 Alteromonas and 14 phages. (b) Phylogenetic analysis of TerL amino acid sequences of 60 phages. (c) The genome-wide tree based on the average nucleotide identity (ANI) from 10 phages. Numbers at the nodes indicate bootstrap values (1000 replications and values >50%).

Figure 4.
The predicted structures of putative receptor binding proteins in vB_AmeP-R8W (R8W). (a) Gp45, putative long tail fibers. (b) Gp34, putative short tail fibers. N-terminal is colored blue, and C-terminal is colored red. The C-score of two proterins are −1.85 and −0.86, respectively.
Figure 4.
The predicted structures of putative receptor binding proteins in vB_AmeP-R8W (R8W). (a) Gp45, putative long tail fibers. (b) Gp34, putative short tail fibers. N-terminal is colored blue, and C-terminal is colored red. The C-score of two proterins are −1.85 and −0.86, respectively.

Figure 5.
(a) Genome BLAST distance phylogeny (GBDP) tree of 57 virus genomes. Based on nucleotide sequences, the GBDP tree is reconstructed by VICTOR, which used the D6 formula and yielded an average support of 71%. Numbers at the nodes are GBDP pseudo-bootstrap values (100 replications and values >50%). (b) ICTV and OPTSIL clusters at the genus and family levels. Each genus is indicated by a unique shape and color. Background colors indicate the 8 OPTSIL clusters at the family level. (c) G + C content and genome sizes.
Figure 5.
(a) Genome BLAST distance phylogeny (GBDP) tree of 57 virus genomes. Based on nucleotide sequences, the GBDP tree is reconstructed by VICTOR, which used the D6 formula and yielded an average support of 71%. Numbers at the nodes are GBDP pseudo-bootstrap values (100 replications and values >50%). (b) ICTV and OPTSIL clusters at the genus and family levels. Each genus is indicated by a unique shape and color. Background colors indicate the 8 OPTSIL clusters at the family level. (c) G + C content and genome sizes.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Statistics of reported genomic information of alterophages.
| Phage Isolate | Isolation Host | Family | Size (bp) | Number of Genes | GC (%) | GenBank |
|---|---|---|---|---|---|---|
| vB_AmeP-R8W | A. mediterranea DE | Autographiviridae | 48,825 | 55 | 40.6 | MW043865 |
| vB_AspP-H4/4 | A. addita H4 | Autographiviridae | 47,631 | 49 | 40.8 | MF278336 |
| vB_AmaP_AD45-P1 | A. macleodii AD45 | Podoviridae | 103,910 | 129 | 43.2 | KF005317 |
| vB_AmaP_AD45-P2 | A. macleodii AD45 | Podoviridae | 104,036 | 129 | 43.2 | KF005320 |
| vB_AmaP_AD45-P3 | A. macleodii AD45 | Podoviridae | 101,724 | 124 | 43.2 | KF005318 |
| vB_AmaP_AD45-P4 | A. macleodii AD45 | Podoviridae | 100,619 | 122 | 43.2 | KF005319 |
| ZP6 | A. macleodii sp. | Podoviridae | 37,743 | 46 | 50.1 | MK203850 |
| PB15 | A. gracilis B15 | Siphoviridae | 37,333 | 61 | 45.5 | KX982260 |
| JH01 | A. marina SW-47 | Siphoviridae | 46,500 | 58 | 44.4 | MH445500 |
| P24 | A. macleodii sp. | Siphoviridae | 46,945 | 74 | 43.8 | MK241539 |
| XX1924 | A. litorea TF-22 | Siphoviridae | 40,580 | 64 | 43.7 | MN592896 |
| vB_AcoS-R7M | A. confluentis DSSK2-12 | Siphoviridae | 56,163 | 67 | 45.6 | MT345684 |
| vB_AmeM_PT11-V22 | A. mediterranea PT11 | Myoviridae | 92,760 | 156 | 38.4 | MN877442 |
Table 2.
The host range of vB_AmeP-R8W (R8W).
| Strain | Isolated From * | Depth | PFU/mL | |||
|---|---|---|---|---|---|---|
| 1011 | 109 | 107 | 105 | |||
| Alteromonas macleodii ATCC 27126T | Hawaii, Pacific Ocean Oahu | Surface seawaters | + | + | + | + |
| Alteromonas marina SW-47T | Eastern Sea, Korea | Surface seawaters | + | + | + | + |
| Alteromonas stellipolaris ANT 69aT | Antarctica | Surface seawaters | + | + | + | + |
| Alteromonas macleodii AD45 | Mediterranean Sea | Surface seawaters | + | + | + | + |
| Alteromonas addita R10SW13T | Chazhma Bay, Sea of Japan, Pacific Ocean | Surface seawaters | + | + | + | + |
| Alteromonas macleodii MCCC 1K00172 | South China Sea | Surface seawaters | + | + | − | − |
| Alteromonas macleodii MCCC 1K00560 | Eastern Pacific Ocean | Surface seawaters | + | + | − | − |
| Alteromonas macleodii AD037 | Port Dickson, Malaysia | Surface seawaters | + | + | − | − |
| Alteromonas macleodii BSH94-8 | Black Sea Karadag | Surface seawaters | + | + | − | − |
| Alteromonas macleodii AD006 | Port Dickson, Malaysia | Surface seawaters | + | + | − | − |
| Alteromonas macleodii MCCC 1K01332 | East Pacific Ocean | Surface seawaters | + | − | − | − |
| Alteromonas confluentis DSSK2-12T | Jeju Island, South Korea | Surface seawaters | + | − | − | − |
| Alteromonas mediterranea EC615 | English Channel | Surface seawaters | + | − | − | − |
| Alteromonas macleodii MCCC 1K00460 | Western Pacific Ocean | Surface seawaters | − | − | − | − |
| Alteromonas macleodii MCCC 1K01839 | Western Pacific Ocean | Surface seawaters | − | − | − | − |
| Alteromonas macleodii MCCC 1K01358 | Eastern Pacific Ocean | Surface seawaters | − | − | − | − |
| Alteromonas macleodii MCCC 1K00811 | South China Sea | Surface seawaters | − | − | − | − |
| Alteromonas australica H 17T | Port Phillip Bay, Tasman Sea, Pacific Ocean | Surface seawaters | − | − | − | − |
| Alteromonas tagae AT1T | Er-Jen River estuary, Tainan | Surface estuarine waters | − | − | − | − |
| Alteromonas lipolytica JW12T | Arabian Sea, Indian Ocean | Surface seawaters | − | − | − | − |
| Alteromonas macleodii BS11 | Black Sea Karadag | Surface seawaters | − | − | − | − |
| Alteromonas mediterranea MED64 | Aegean Sea, Mediterranean | Surface seawaters | − | − | − | − |
| Alteromonas macleodii BS7 | Black Sea Karadag | Surface seawaters | − | − | − | − |
| Alteromonas macleodii EC673 | English Channel | Surface seawaters | − | − | − | − |
| Alteromonas alba 190T | Western Pacific Ocean | Surface seawaters | − | − | − | − |
| Alteromonas macleodii BSH84-3 | Black Sea Karadag | Surface seawaters | − | − | − | − |
| Alteromonas macleodii BS8 | Black Sea Karadag | Surface seawaters | − | − | − | − |
| Alteromonas simiduii AS1T | Er-Jen River estuary, Tainan | Surface estuarine waters | − | − | − | − |
| Alteromonas macleodii MCCC 1K01842 | Western Pacific Ocean | Subsurface seawaters (75 m) | + | + | − | − |
| Alteromonas macleodii MCCC 1K01832 | Western Pacific Ocean | Subsurface seawaters (30 m) | + | − | − | − |
| Alteromonas macleodii MCCC 1K01840 | Western Pacific Ocean | Subsurface seawater (30 m) | + | − | − | − |
| Alteromonas macleodii MCCC 1K01294 | Western Pacific Ocean | Subsurface seawaters (75 m) | + | − | − | − |
| Alteromonas macleodii MCCC 1K01823 | Western Pacific Ocean | Subsurface seawaters (75 m) | − | − | − | − |
| Alteromonas macleodii A16(2794) | South China Sea | Subsurface seawater (75 m) | − | − | − | − |
| Alteromonas macleodii A14(2783) | South China Sea | Subsurface seawaters (75 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K01274 | Western Pacific Ocean | Subsurface seawaters (100 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K01826 | Western Pacific Ocean | Subsurface seawaters (100 m) | − | − | − | − |
| Alteromonas mediterranea DET | Adriatic Sea, Urania Basin | Deep seawaters (1000 m) | + | + | + | + |
| Alteromonas macleodii MCCC 1A04487 | Northwestern Pacific Ocean | Deep seawaters (2700 m) | + | + | + | − |
| Alteromonas mediterranea DE1 | Adriatic Sea, Urania Basin | Deep seawaters (1000 m) | + | + | − | − |
| Alteromonas macleodii MCCC 1A07993 | Southern Atlantic Ocean | Deep seawaters (2147 m) | + | + | − | − |
| Alteromonas macleodii MCCC 1A09262 | Southern Atlantic Ocean | Deep seawaters (3047 m) | + | + | − | − |
| Alteromonas mediterranea UM7 | Ionian Sea, Uranian Basin Western of Crete | Deep seawaters (3475 m) | + | + | − | − |
| Alteromonas mediterranea UM8 | Ionian Sea, Uranian Basin Western of Crete | Deep seawaters (3475 m) | + | + | − | − |
| Alteromonas macleodii MCCC 1A00323 | Atlantic Ocean | Deep seawaters (3542 m) | + | + | − | − |
| Alteromonas macleodii MCCC 1K02087 | South China Sea | Deep seawaters (1700 m) | + | − | − | − |
| Alteromonas macleodii MCCC 1K00565 | Eastern Pacific Ocean | Deep seawaters (5098 m) | + | − | − | − |
| Alteromonas macleodii MCCC 1K00800 | Eastern Pacific Ocean | Deep seawaters (1000 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1A02046 | Indian Ocean | Deep seawaters (2391 m) | − | − | − | − |
| Alteromonas mediterranea UM4b | Ionian Sea, Uranian Basin Western of Crete | Deep seawaters (3455 m) | − | − | − | − |
| Alteromonas mediterranea U4 | Ionian Sea, Uranian Basin Western of Crete | Deep seawaters (3475 m) | − | − | − | − |
| Alteromonas mediterranea U7 | Ionian Sea, Uranian Basin Western of Crete | Deep seawaters (3500 m) | − | − | − | − |
| Alteromonas mediterranea U8 | Ionian Sea, Uranian Basin Western of Crete | Deep seawaters (3500 m) | − | − | − | − |
| Alteromonas macleodii U12 | Ionian Sea, Uranian Basin Western of Crete | Deep seawaters (3500 m) | − | − | − | − |
| Alteromonas macleodii A25 | South China Sea | Deep seawaters (4058 m) | − | − | − | − |
| Alteromonas macleodii A27 | South China Sea | Deep seawaters (4058 m) | − | − | − | − |
| Alteromonas sp. MCCC 1A07988 | Southern Atlantic Ocean | Deep seawaters (5610 m) | − | − | − | − |
| Alteromonas gracilis 9a2T | Pacific Ocean | Sediment (6310 m) | + | + | + | + |
| Alteromonas sp. MCCC 1A09157 | Southern Atlantic Ocean | Sediment | + | + | − | − |
| Alteromonas naphthalenivorans SN2T | Taean, South Korea | Sediment (tidal-flat) | + | + | − | − |
| Alteromonas macleodii MCCC 1K02779 | Atlantic Ocean | Sediment (2577 m) | + | − | − | − |
| Alteromonas sp. MCCC 1A09130 | Southern Atlantic Ocean | Sediment | + | − | − | − |
| Alteromonas macleodii MCCC 1K02456 | Northwestern Indian Ocean | Sediment (1818 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K02451 | Northwestern Indian Ocean | Sediment (2009 m) | − | − | − | − |
| Alteromonas sp. MCCC 1A08050 | Southern Atlantic Ocean | Sediment (2481 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K02444 | Northwestern Indian Ocean | Sediment (2540 m) | − | − | − | − |
| Alteromonas pelagimontana 5.12T | Indian Ocean | Sediment (2681 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K01703 | Atlantic Ocean | Sediment (2781m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K01716 | Atlantic Ocean | Sediment (2781 m) | − | − | − | − |
| Alteromonas litorea TF-22T | Korea, Yellow Sea | Sediment (Intertidal) | − | − | − | − |
| Alteromonas aestuariivivens JDTF-113 | Jindo, South Korea | Sediment (tidal-flat) | − | − | − | − |
| Alteromonas sp. EZ55 | Tropical Pacific Ocean | Prochlorococcus culture (20 m) | + | + | + | + |
| Alteromonas macleodii MCCC 1F01223 | Xiamen, China | Algae culture | + | + | − | − |
| Alteromonas hispanica F-32T | Fuente de Piedra, southern Spain | Hypersaline water | + | + | − | − |
| Alteromonas genovensis LMG 24078T | Genoa, Italy | Biofilm | − | − | − | − |
| Alteromonas macleodii MCCC 1K02452 | Northwestern Indian Ocean | Olivine (3042 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K00767 | Eastern Pacific Ocean | Seawaters (500 m) | − | − | − | − |
| Alteromonas macleodii MCCC 1K01276 | Western Pacific Ocean | Seawaters (300 m) | − | − | − | − |
| Alteromonas macleodii AS7 | Andaman Sea | NA | − | − | − | − |
* The strains isolated from the same sea area may be isolated from different stations. Please refer to the attachment for Supplementary Materials Table S1.
Table 3.
RBPs of alterophages.
| Phage | #Accession | aa Length | NCBI Annotation | HHpred (Probability|E-Value|Aligned Cols|Identities) | Phyre2 (Confidence|Identity) | β-Helix |
|---|---|---|---|---|---|---|
| vB_AmeP-R8W | - | 156 | tail fiber protein | pectin degradation protein (99.11%|5.3 × 10−9|94|13%) | dimethylsulfoniopropionate lyase (92.8%|18%) | YES |
| vB_AmeP-R8W | - | 1492 | tail fiber protein | glycoside hydrolase (96.68%|0.062|171|18%) | hydrolase,sialidase (91.3%|27%) | YES |
| vB_AspP-H4/4 | ASL24413 | 157 | tail fiber protein | pectin degradation protein (99.12%|4.6 × 10−9|83|19%) | dimethylsulfoniopropionate lyase (92.9%|18%) | YES |
| vB_AspP-H4/4 | ASL24424 | 1524 | tail fiber protein | glycoside hydrolase (97.14%|0.021|187|19%) | hydrolase,sialidase (91.3%|27%) | YES |
| Prokaryotic dsDNA virus TS | AWN07083 | 777 | tail fiber protein | pectin degradation protein (99.23%|7.7 × 10−10|83|18%) | dimethylsulfoniopropionate lyase (92.3%|18%) | YES |
| Prokaryotic dsDNA virus TS | QDP58699 | 954 | tail fiber protein | tail fiber protein (99.82%|6.6 × 10−18|278|14%) | tailspike gp27 (60.5%|23%) | YES |
| ZP6 | AZS06567 | 873 | tailspike protein | phiAB6 tailspike (99.92%|3.5 × 10−22|402|13%) | alpha-1,3-glucanase (99.95%|14%) | YES |
| ZP6 | QMS42070 | 629 | tailspike protein | tailspike protein (99.92%|4.8 × 10−22|383|14%) | glucan 1,3-beta-glucosidase (96.9%|26%) | YES |
| vB_AmaP_AD45-P1 | AGM46838 | 1545 | tail fiber protein | endo-N-acetylneuraminidase (98.09%|0.000014|95|21%) | chaperone,endo-N-acetylneuraminidase (99.1%|18%) | YES |
| vB_AmaP_AD45-P1 | AGM46839 | 1236 | tail fiber protein | NF | long-tail fiber (84.8%|11%) | YES |
| vB_AmaP_AD45-P2 | AGM47190 | 1545 | tail fiber protein | endo-N-acetylneuraminidase (98.09%|0.000014|95|21%) | chaperone,endo-N-acetylneuraminidase (99.1%|18%) | YES |
| vB_AmaP_AD45-P2 | AGM47191 | 1236 | tail fiber protein | NF | long-tail fiber (84.8%|11%) | YES |
| vB_AmaP_AD45-P3 | AGM46957 | 1545 | tail fiber protein | endo-N-acetylneuraminidase (97.14%|0.000014|57|18%) | chaperone,endo-N-acetylneuraminidase (99.1%|18%) | YES |
| vB_AmaP_AD45-P4 | AGM46958 | 1236 | tail fiber protein | NF | long-tail fiber (84.8%|11%) | YES |
| vB_AmaP_AD45-P4 | AGM47074 | 1545 | tail fiber protein | endo-N-acetylneuraminidase (98.09%|0.000014|95|21%) | chaperone,endo-N-acetylneuraminidase (99.1%|18%) | YES |
| vB_AmaP_AD45-P4 | AGM47075 | 1236 | tail fiber protein | NF | long-tail fiber (84.8%|11%) | YES |
| PB15 | APC46581 | 748 | tailspike protein | particle-associated lyase (99.86%|3.1 × 10−18|328|14%) | hydrolase, xylosidase (83.9%|25%) | YES |
| PB15 | APC46582 | 731 | tail fiber protein | endo-beta-N-acetylglucosaminidase (97.08%| 0.019|119|5%) | hydrolase, spgh29 (98.4%|18%) | YES |
| JH01 | AWY02808 | 112 | capsid fiber protein | Capsid fiber protein (64.4|24|238|347) alpha-galactosidase (99.35%|5.4 × 10−11|93|14%) | altronate hydrolase (53.4%|20%) | NO |
| XX1924 | QGZ13097 | 518 | discoidin domain-containing protein | endo-beta-N-acetylglucosaminidase (95.448%| 0.016|119|5%) | hydrolase, spgh29 (96.5%|18%) | YES |
| XX1924 | QGZ13160 | 352 | tail fiber protein | tailspike protein (97.81%|0.0075|212|11%) | antimicrobial protein, neutrophil defensin 4 (51.6%|62%) | NO |
| P24 | AZU97343 | 361 | tail fiber protein | NF | nf | |
| vB_AcoS-R7M | YP_009859590 | 273 | ribonuclease III | NF | sugar binding protein (47%|10%) | NO |
| vB_AmeM_PT11-V22 | QHZ59724 | 369 | tail fiber protein | - | - | - |
NF = not found.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Feng, X.; Yan, W.; Wang, A.; Ma, R.; Chen, X.; Lin, T.-H.; Chen, Y.-L.; Wei, S.; Jin, T.; Jiao, N.; et al. A Novel Broad Host Range Phage Infecting Alteromonas. Viruses 2021, 13, 987. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060987
AMA Style
Feng X, Yan W, Wang A, Ma R, Chen X, Lin T-H, Chen Y-L, Wei S, Jin T, Jiao N, et al. A Novel Broad Host Range Phage Infecting Alteromonas. Viruses. 2021; 13(6):987. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060987
Chicago/Turabian StyleFeng, Xuejin, Wei Yan, Anan Wang, Ruijie Ma, Xiaowei Chen, Ta-Hui Lin, Yi-Lung Chen, Shuzhen Wei, Tao Jin, Nianzhi Jiao, and et al. 2021. "A Novel Broad Host Range Phage Infecting Alteromonas" Viruses 13, no. 6: 987. https://0-doi-org.brum.beds.ac.uk/10.3390/v13060987
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.