
Impact of Analytical Treatment Interruption on Burden and Diversification of HIV Peripheral Reservoir: A Pilot Study

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Total HIV-DNA and Residual Viremia Quantification
2.3. Viral Sequencing
2.4. Estimation of Major Drug Resistance Mutations and Hypermutated Pol Sequences
2.5. Phylogenetic Analysis and Genealogical Sorting Index (GSI)
3. Results
3.1. Patients’ Population
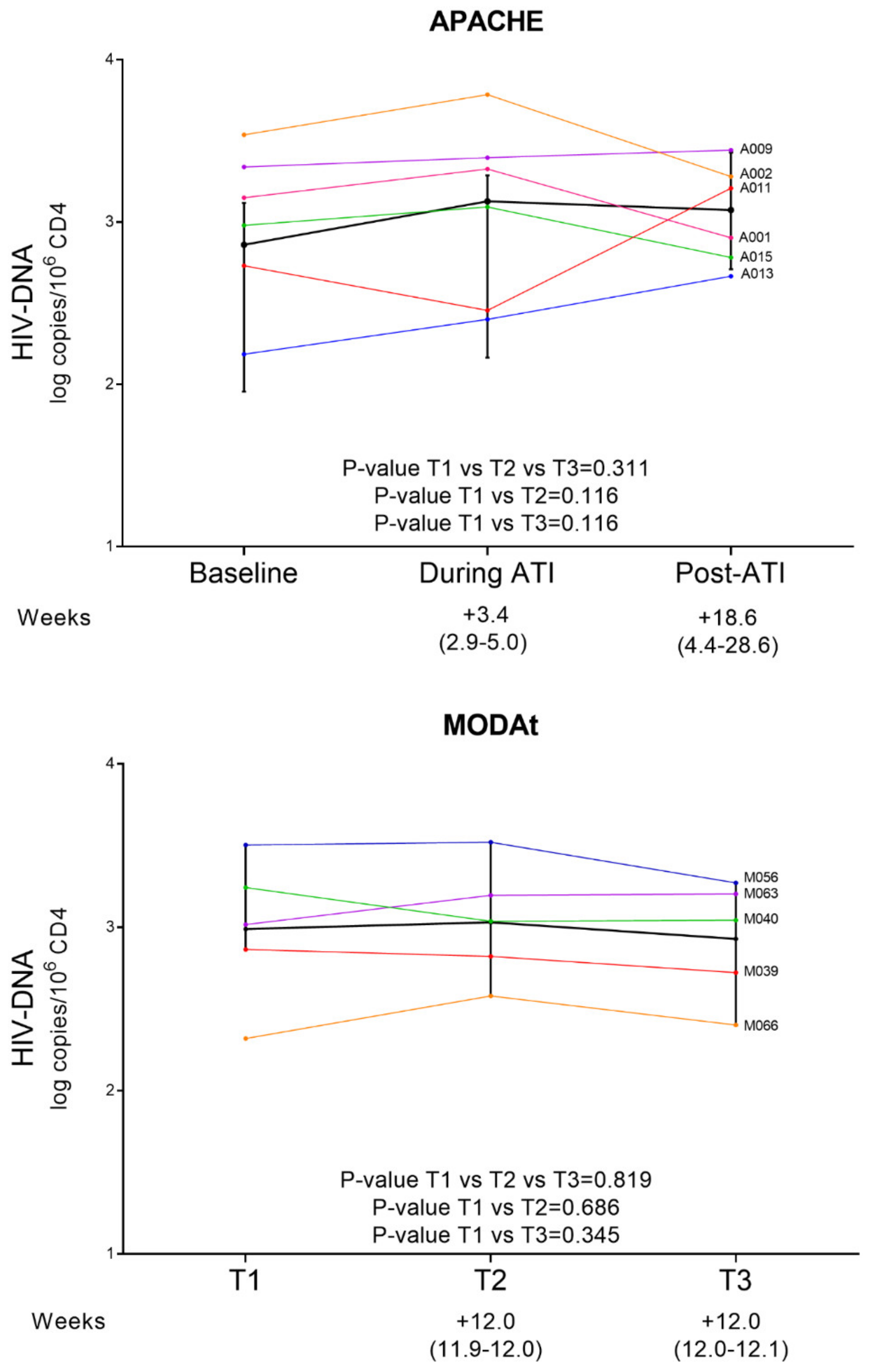
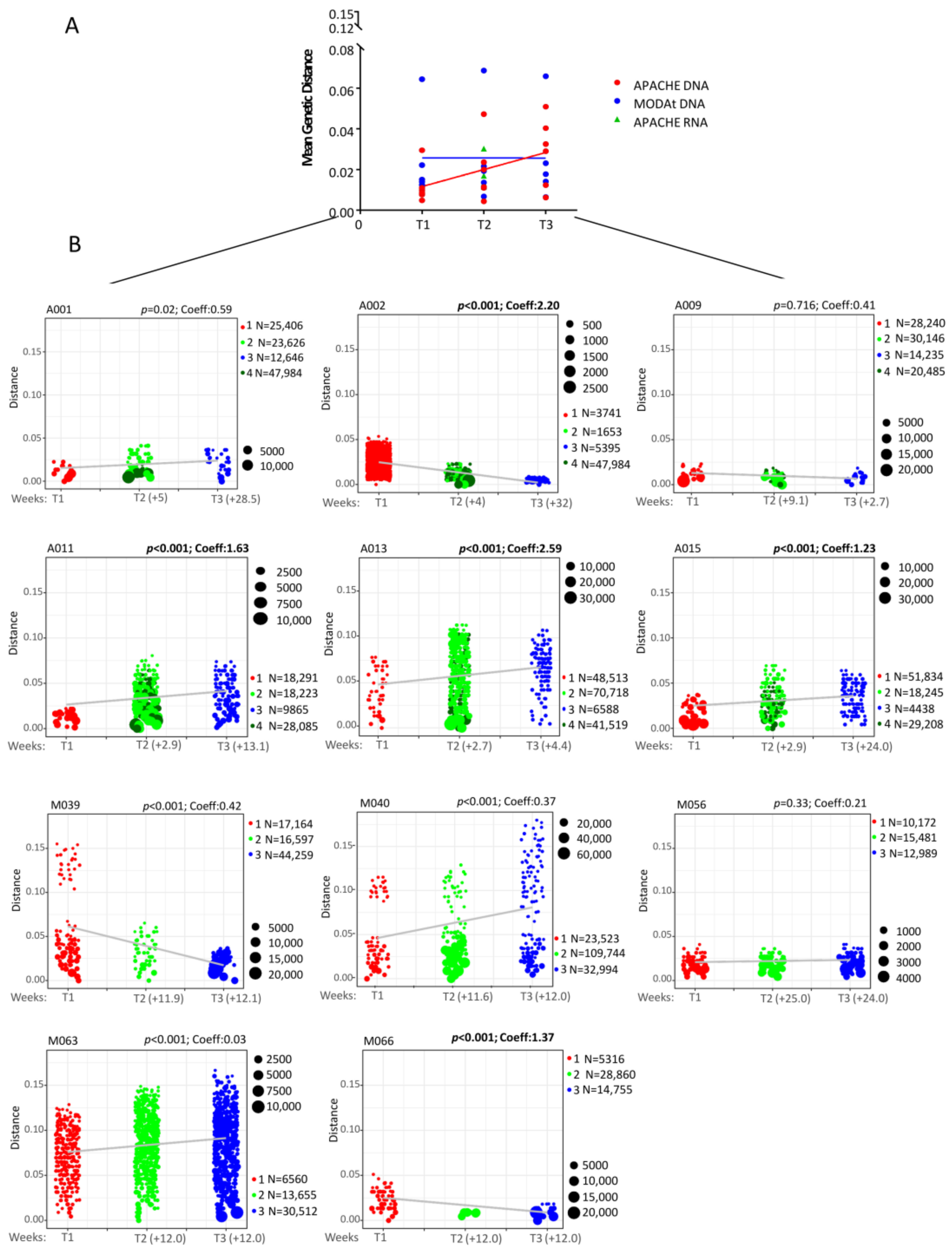
3.2. Quantitative Measures of Reservoir Change
3.3. Major Drug Resistance Mutations Prevalence According to Time Points
3.4. Defective Mutants over Time
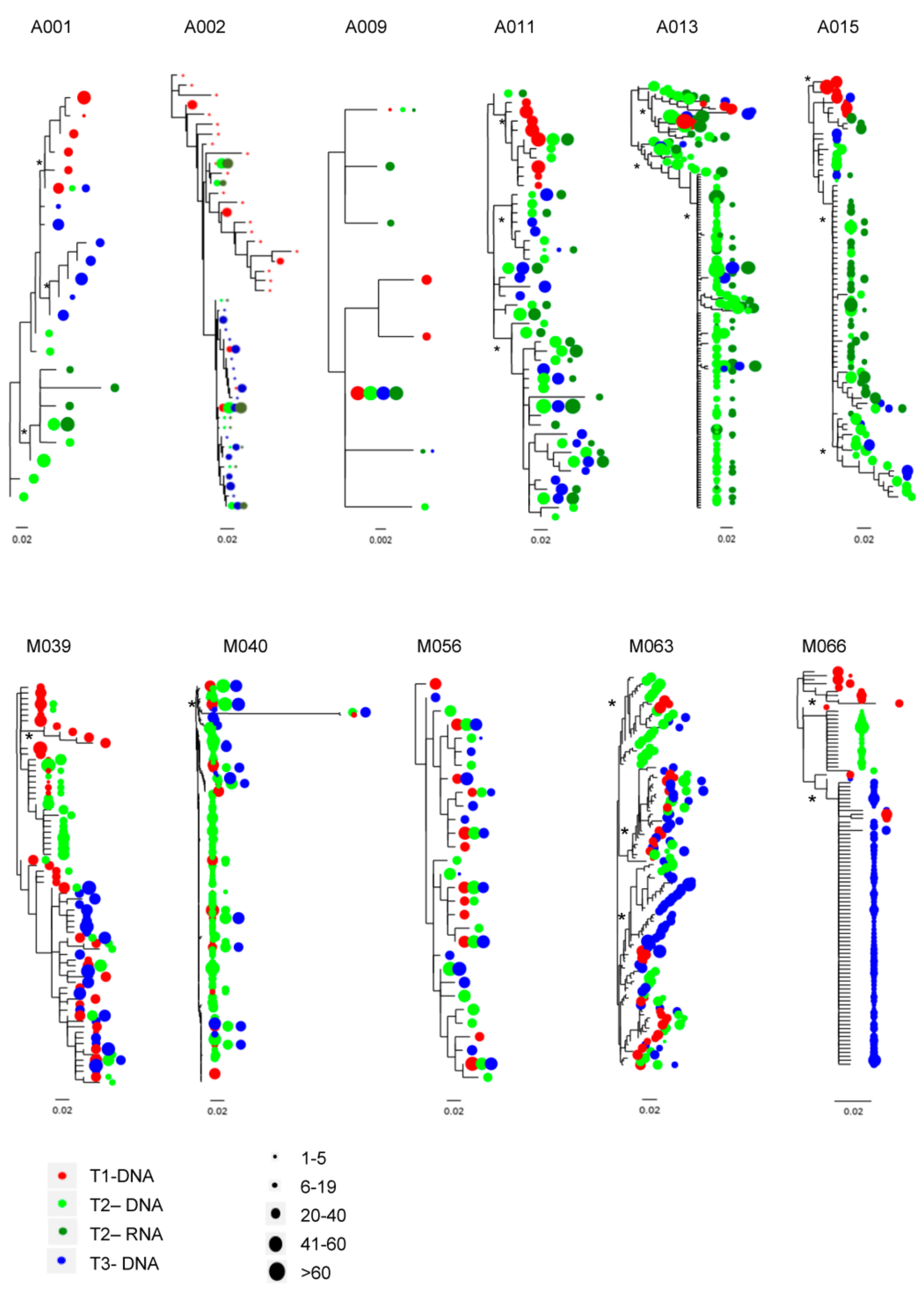
3.5. C2-V3-C3 Evolution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perelson, A.S.; Essunger, P.; Cao, Y.; Vesanen, M.; Hurley, A.; Saksela, K.; Markowitz, M.; Ho, D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 1997, 387, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Michaels, S.H.; Clark, R.; Kissinger, P. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 1998, 339, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Besson, G.J.; Lalama, C.M.; Bosch, R.J.; Gandhi, R.T.; Bedison, M.A.; Aga, E.; Riddler, S.A.; McMahon, D.K.; Hong, F.; Mellors, J.W. HIV-1 DNA decay dynamics in blood during more than a decade of suppressive antiretroviral therapy. Clin. Infect Dis. 2014, 59, 1312–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alteri, C.; Scutari, R.; Stingone, C.; Maffongelli, G.; Brugneti, M.; Falasca, F.; Martini, S.; Bertoli, A.; Turriziani, O.; Sarmati, L.; et al. Quantification of HIV-DNA and residual viremia in patients starting ART by droplet digital PCR: Their dynamic decay and correlations with immunological parameters and virological success. J. Clin. Virol. 2019, 117, 61–67. [Google Scholar] [CrossRef]
- Golob, J.L.; Stern, J.; Holte, S.; Kitahata, M.M.; Crane, H.M.; Coombs, R.W.; Goecker, E.; Woolfrey, A.E.; Harrington, R.D. HIV DNA levels and decay in a cohort of 111 long-term virally suppressed patients. AIDS 2018, 32, 2113–2118. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hanhauser, E.; Hu, Z.; Stellbrink, H.J.; Noah, C.; Martin, J.N.; Deeks, S.G.; Kuritzkes, D.R.; Pereyra, F. Viremic control and viral coreceptor usage in two HIV-1-infected persons homozygous for CCR5 Δ32. AIDS 2015, 29, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 2014, 161, 319–327. [Google Scholar] [CrossRef]
- Katlama, C.; Lambert-Niclot, S.; Assoumou, L.; Papagno, L.; Lecardonnel, F.; Zoorob, R.; Tambussi, G.; Clotet, B.; Youle, M.; Achenbach, C.J.; et al. Treatment intensification followed by interleukin-7 reactivates HIV without reducing total HIV DNA: A randomized trial. AIDS 2016, 30, 221–230. [Google Scholar] [CrossRef]
- Bar, K.J.; Sneller, M.C.; Harrison, L.J.; Justement, J.S.; Overton, E.T.; Petrone, M.E.; Salantes, D.B.; Seamon, C.A.; Scheinfeld, B.; Kwan, R.W.; et al. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med. 2016, 375, 2037–2050. [Google Scholar] [CrossRef]
- Yerly, S.; Kaiser, L.; Perneger, T.V.; Cone, R.W.; Opravil, M.; Chave, J.P.; Furrer, H.; Hirschel, B.; Perrin, L. Time of initiation of antiretroviral therapy: Impact on HIV-1 viraemia. The Swiss HIV Cohort Study. AIDS 2000, 14, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.; Magierowska, M.; Royz, M.; Foley, B.; Peddada, L.; Smith, R.; Heldebrant, C.; Conrad, A.; Busch, M. Homogeneous quasispecies in 16 out of 17 individuals during very early HIV-1 primary infection. AIDS 2002, 16, 189–195. [Google Scholar] [CrossRef]
- Oxenius, A.; Price, D.A.; Easterbrook, P.J.; O’Callaghan, C.A.; Kelleher, A.D.; Whelan, J.A.; Sontag, G.; Sewell, A.K.; Phillips, R.E. Early highly active antiretroviral therapy for acute HIV-1 infection preserves immune function of CD8+ and CD4+ T lymphocytes. Proc. Natl. Acad. Sci USA 2000, 97, 3382–3387. [Google Scholar] [CrossRef] [Green Version]
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Cockerham, L.R.; Hatano, H.; Deeks, S.G. Post-Treatment Controllers: Role in HIV ”Cure” Research. Curr. HIV/AIDS Rep. 2016, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Volberding, P.; Demeter, L.; Bosch, R.J.; Aga, E.; Pettinelli, C.; Hirsch, M.; Vogler, M.; Martinez, A.; Little, S.; Connick, E.; et al. Antiretroviral therapy in acute and recent HIV infection: A prospective multicenter stratified trial of intentionally interrupted treatment. AIDS 2009, 23, 1987–1995. [Google Scholar] [CrossRef] [Green Version]
- Hocqueloux, L.; Prazuck, T.; Avettand-Fenoel, V.; Lafeuillade, A.; Cardon, B.; Viard, J.P.; Rouzioux, C. Long-term immunovirologic control following antiretroviral therapy interruption in patients treated at the time of primary HIV-1 infection. AIDS 2010, 24, 1598–1601. [Google Scholar] [CrossRef]
- Calin, R.; Hamimi, C.; Lambert-Niclot, S.; Carcelain, G.; Bellet, J.; Assoumou, L.; Tubiana, R.; Calvez, V.; Dudoit, Y.; Costagliola, D.; et al. Treatment interruption in chronically HIV-infected patients with an ultralow HIV reservoir. AIDS 2016, 30, 761–769. [Google Scholar] [CrossRef]
- Fehér, C.; Plana, M.; Crespo Guardo, A.; Climent, N.; Leal, L.; Ugarte, A.; Fernández, I.; Etcheverry, M.F.; Gatell, J.M.; Sánchez-Palomino, S.; et al. Classifier to Predict Viral Control After Antiretroviral Treatment Interruption in Chronic HIV-1-Infected Patients. J. Acquir. Immune. Defic. Syndr. 2020, 83, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarridge, K.E.; Blazkova, J.; Einkauf, K.; Petrone, M.; Refsland, E.W.; Justement, J.S.; Shi, V.; Huiting, E.D.; Seamon, C.A.; Lee, G.Q.; et al. Effect of analytical treatment interruption and reinitiation of antiretroviral therapy on HIV reservoirs and immunologic parameters in infected individuals. PLoS Pathog. 2018, 14, e1006792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salantes, D.B.; Zheng, Y.; Mampe, F.; Srivastava, T.; Beg, S.; Lai, J.; Li, J.Z.; Tressler, R.L.; Koup, R.A.; Hoxie, J.; et al. HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J. Clin. Investig. 2018, 128, 3102–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggiolo, F.; di Filippo, E.; Comi, L.; Callegaro, A. Post-treatment controllers after treatment interruption in chronically HIV-infected patients. AIDS 2018, 32, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.S.Y.; Smith, M.Z.; Lewin, S.R.; McMahon, J.H. Clinical trials of antiretroviral treatment interruption in HIV-infected individuals. AIDS 2019, 33, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Immonen, T.T.; Fennessey, C.M.; Lipkey, L.; Thorpe, A.; del Prete, G.Q.; Lifson, J.D.; Davenport, M.P.; Keele, B.F. Transient viral replication during analytical treatment interruptions in SIV infected macaques can alter the rebound-competent viral reservoir. PLoS Pathog. 2021, 17, e1009686. [Google Scholar] [CrossRef] [PubMed]
- Castagna, A.; Muccini, C.; Galli, L.; Bigoloni, A.; Poli, A.; Spagnuolo, V.; Nozza, S.; Racca, S.; Galli, A.; Cinque, P.; et al. Analytical treatment interruption in chronic HIV-1 infection: Time and magnitude of viral rebound in adults with 10 years of undetectable viral load and low HIV-DNA (APACHE study). J. Antimicrob. Chemother. 2019, 74, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Julg, B.; Dee, L.; Ananworanich, J.; Barouch, D.H.; Bar, K.; Caskey, M.; Colby, D.J.; Dawson, L.; Dong, K.L.; Dubé, K.; et al. Recommendations for analytical antiretroviral treatment interruptions in HIV research trials-report of a consensus meeting. Lancet HIV 2019, 6, e259–e268. [Google Scholar] [CrossRef]
- Galli, L.; Spagnuolo, V.; Bigoloni, A.; D’Arminio Monforte, A.; Montella, F.; Antinori, A.; di Biagio, A.; Rusconi, S.; Guaraldi, G.; di Giambenedetto, S.; et al. MODAt Study Group. Atazanavir/ritonavir monotherapy: 96 week efficacy, safety and bone mineral density from the MODAt randomized trial. J. Antimicrob. Chemother. 2016, 71, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtgast, E.J.; Sima, V.M.; Bertels, K.; Al-Ars, Z. Hardware acceleration of BWA-MEM genomic short read mapping for longer read lengths. Comput Biol. Chem. 2018, 75, 54–64. [Google Scholar] [CrossRef]
- Töpfer, A.; Zagordi, O.; Prabhakaran, S.; Roth, V.; Halperin, E.; Beerenwinkel, N. Probabilistic inference of viral quasispecies subject to recombination. J. Comput Biol. 2013, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Fourati, S.; Lambert-Niclot, S.; Soulie, C.; Malet, I.; Valantin, M.A.; Descours, B.; Ait-Arkoub, Z.; Mory, B.; Carcelain, G.; Katlama, C.; et al. HIV-1 genome is often defective in PBMCs and rectal tissues after long-term HAART as a result of APOBEC3 editing and correlates with the size of reservoirs. J. Antimicrob. Chemother. 2012, 67, 2323–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.P.; Korber, B.T. Detecting hypermutations in viral sequences with an emphasis on G --> A hypermutation. Bioinformatics 2000, 16, 400–401. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Cummings, M.P.; Neel, M.C.; Shaw, K.L. A genealogical approach to quantifying lineage divergence. Evolution 2008, 62, 2411–2422. [Google Scholar] [CrossRef]
- Haynes, W. Benjamini–Hochberg Method. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.R.; et al. International AIDS Society global scientific strategy: Towards an HIV cure 2016. Nat. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef]
- Pitman, M.C.; Lau, J.S.Y.; McMahon, J.H.; Lewin, S.R. Barriers and strategies to achieve a cure for HIV. Lancet HIV 2018, 5, e317–e328. [Google Scholar] [CrossRef]
- Avettand-Fènoël, V.; Boufassa, F.; Galimand, J.; Meyer, L.; Rouzioux, C. ANRS SEROCO Cohort Study Group. HIV-1 DNA for the measurement of the HIV reservoir is predictive of disease progression in seroconverters whatever the mode of result expression is. J. Clin. Virol. 2008, 42, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, R.; Lee, G.Q.; Sun, X.; Etemad, B.; Aboukhater, L.M.; Hu, Z.; Brumme, Z.L.; Aga, E.; Bosch, R.J.; Wen, Y.; et al. HIV-1 proviral landscapes distinguish posttreatment controllers from noncontrollers. J. Clin. Investig. 2018, 128, 4074–4085. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patients’ Characteristics | Overall (N = 11) | APACHE Study (N = 6) | MODAt Study (N = 5) | p-Value § |
|---|---|---|---|---|
| Age (years), median (IQR) | 47.3 (43.4–1.2) | 49.5 (47.3–51.2) | 44.1 (43.4–45.2) | 0.235 |
| Male gender, N (%) | 8 (72.7) | 5 (83.3) | 3 (60.0) | 0.545 |
| Subtype B, N (%) | 8 (72.7) | 3 (50.0) | 5 (100.0) | 0.182 |
| Years since first HIV positive test, median (IQR) | 15.3 (9.3–19.4) | 17.1 (15.3–22.8) | 9.3 (4.0–11.4) | 0.055 |
| Years since ART start, median (IQR) | 11.1 (6.1–17.6) | 17.1 (13.5–18.1) | 6.1 (3.2–9.0) | 0.008 |
| Years of HIV-1 RNA <50 copies/mL, median (IQR) | 9.6 (4.7–11.7) | 11.5 (11.0–11.9) | 4.7 (2.9–6.7) | 0.008 |
| Viro-immunological parameters at T1 (median, IQR): | ||||
| HIV-DNA (copies/106 CD4+T-cells) | 1037 (730–1311) | 1048 (863–1286) | 1037 (730–1750) | 0.792 |
| HIV-RNA * (copies/mL) | 2 (2–7) | 2 (2–5) | 3 (2–7) | 0.662 |
| CD4+T-cells (cells/mm3) | 748 (563–1012) | 737 (563–1512) | 774 (745–777) | 0.931 |
| CD8+T-cells (cells/mm3) | 814 (451–1203) | 1017 (333–1235) | 685 (534–814) | 0.523 |
| CD4+/CD8+ ratio | 1.24 (0.72–2.04) | 1.44 (0.61–2.15) | 1.24 (0.82–1.46) | 0.784 |
| Viro-immunological parameters at T2 (median, IQR): | ||||
| HIV-DNA (copies/106 CD4+T-cells) | 1567 (398–3307) | 1916 (398–3798) | 1087 (663–1567) | 0.537 |
| HIV-RNA (copies/mL) | 2984 (3–69,820) | 60,108 (12,251–1,680,510) | 3 (2–4) * | 0.004 |
| CD4+T-cells (cells/mm3) | 731 (521–1004) | 590 (497–1064) | 765 (731–890) | 0.537 |
| CD8+T-cells (cells/mm3) | 829 (454–1341) | 1041 (279–1341) | 750 (594–1296) | 0.998 |
| CD4+/CD8+ ratio | 1.19 (0.54–1.46) | 1.13 (0.53–1.78) | 1.19 (0.77–1.29) | 0.784 |
| Viro-immunological parameters at T3 (median, IQR): | ||||
| HIV-DNA (copies/106 CD4+T-cells) | 1104 (527–1867) | 1333 (553–2183) | 1104 (527–1598) | 0.537 |
| HIV-RNA * (copies/mL) | 3 (0–7) | 4 (1–9) | 0 (0–5) | 0.329 |
| CD4+T-cells (cells/mm3) | 816 (645–1096) | 987 (645–1134) | 728 (728–816) | 0.429 |
| CD8+T-cells (cells/mm3) | 1003 (523–1257) | 1066 (491–2058) | 1003 (744–1043) | 0.783 |
| CD4+/CD8+ ratio | 0.87 (0.62–1.33) | 1.06 (0.51–1.33) | 0.85 (0.78–0.98) | 0.927 |
| Study | ID | Time 1 | Time 2 | Time 3 | |
|---|---|---|---|---|---|
| APACHE | Sample | HIV-DNA | HIV-DNA | HIV-RNA | HIV-DNA |
| A001 | PR: None RT: D67N (99.4%), K70R (99.6%), M184V (14.0%), T215I (85.6%), K219Q (99.8%) | PR: None RT: D67N (21.3%), K70R (99.7%), M184V (11.1%), K219Q (10.2%) | PR: None RT: K70R (99.6%) | PR: None RT: D67N (99.3%), K70R (99.7%), M184V (8.1%), T215I (91.9%), K219Q (99.8%) | |
| A002 | PR: None RT: W88 *(5.0%), G190E (5.1%), W212 *(5.0%) | PR: None RT: W88 *(5.0%), W212 *(7.0%) | PR: None RT: W88 *(5.0%), W212 *(7.0%) | PR: None RT: None | |
| A009 | PR: None RT: D67N (6.2%), E138K (7.3%), W153 * (4.8%), W212 * (8.5%) | PR: None RT: None | PR: None RT: None | PR: None RT: None | |
| A011 | PR: None RT: None | PR: None RT: None | PR: None RT: None | PR: None RT: None | |
| A013 | PR: None RT: None | PR: None RT: None | PR: None RT: None | PR: None RT: None | |
| A015 | PR: None RT: K65R (39.5%), K101E (38.0%), Y181C (40.2%), M184I (28.9%) | PR: None RT: Q23 * (71.0%), K65R (3.5%), Y181C (6.5%), M184I (3.6%), W153 *(5.0%) | PR: None RT: None | PR: None RT: None | |
| MODAt | Sample | HIV-DNA | HIV-DNA | HIV-DNA | |
| M039 | PR: None RT: M41L (99.6%), T215C (99.5%) | PR: None RT: M41L (99.6%), M184I (6.9%), T215C (99.4%) | PR: None RT: M41L (93.7%), T215C (98.6%) | ||
| M040 | PR: None RT: M41L (47.1%) | PR: None RT: M41L (73.9%) | PR: None RT: M41L (31.7%) | ||
| M056 | PR: None RT: None | PR: None RT: None | PR: None RT: None | ||
| M063 | PR: W42 *(8.0%) RT: None | PR: None RT: None | PR: None RT: None | ||
| M066 | PR: None RT: None | PR: None RT: None | PR: W42 *(5.0%), M46I (5.1%) RT: W153 * (12.1%) | ||
| T1 DNA vs. T2 DNA | T1 DNA vs. T2 RNA | T1 DNA vs. T3 DNA | T2 DNA vs. T2 RNA | T2 RNA vs. T3 DNA | T2 DNA vs. T3 DNA | ||
|---|---|---|---|---|---|---|---|
| APACHE | A001 | 1.00 * vs. 0.99 * | 1.00 * vs. 0.99 * | 0.50 vs. 0.99 * | 0.11 vs. 0.32 | 1.00 * vs. 0.99 * | 0.96 * vs. 0.98 * |
| APACHE | A002 | 0.16 vs. 0.68 * | 0.32 vs. 0.72 * | 0.61 * vs. 0.73 * | 0.43 vs. 0.68 * | 0.01 vs. 0.01 | 0.29 vs. 0.45 |
| APACHE | A009 | 0.20 vs. 0.08 | 0.08 vs. 0.10 | 0.06 vs. 0.08 | 0.05 vs. 0.08 | 0.05 vs. 0.08 | 0.07 vs. 0.18 |
| APACHE | A011 | 0.89 * vs. 0.52 * | 0.93 * vs. 0.57 * | 1.00 * vs. 0.99 * | 0.11 vs. 0.16 | 0.48 vs. 0.44 | 0.42 vs. 0.46 |
| APACHE | A013 | 0.85 * vs. 0.70 * | 0.92 * vs. 0.99 * | 0.94 * vs. 0.56 * | 0.11 vs. 0.26 | 0.31 vs. 0.21 | 0.37 vs. 0.19 |
| APACHE | A015 | 0.99 * vs. 0.84 * | 0.93 * vs. 0.55 * | 0.93 * vs. 0.55 * | 0.28 vs. 0.31 | 0.90 * vs. 0.45 | 0.69 * vs. 0.47 |
| MODAt | M039 | 0.62 * vs. 0.33 | // | 0.47 vs. 0.78 * | // | // | 0.34 vs. 0.81 * |
| MODAt | M040 | 0.10 vs. 0.16 | // | 0.10 vs. 0.18 | // | // | 0.16 vs. 0.18 |
| MODAt | M056 | 0.23 vs. 0.11 | // | 0.11 vs. 0.15 | // | // | 0.20 vs. 0.37 |
| MODAt | M063 | 0.32 vs. 0.40 | // | 0.75 * vs. 0.51 * | // | // | 0.38 vs. 0.37 |
| MODAt | M066 | 1.00 * vs. 0.99 * | // | 1.00 * vs. 0.95 * | // | // | 1.00 * vs. 0.99 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scutari, R.; Costabile, V.; Galli, L.; Bellocchi, M.C.; Carioti, L.; Barbaliscia, S.; Poli, A.; Galli, A.; Perno, C.F.; Santoro, M.M.; et al. Impact of Analytical Treatment Interruption on Burden and Diversification of HIV Peripheral Reservoir: A Pilot Study. Viruses 2021, 13, 1403. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071403
Scutari R, Costabile V, Galli L, Bellocchi MC, Carioti L, Barbaliscia S, Poli A, Galli A, Perno CF, Santoro MM, et al. Impact of Analytical Treatment Interruption on Burden and Diversification of HIV Peripheral Reservoir: A Pilot Study. Viruses. 2021; 13(7):1403. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071403
Chicago/Turabian StyleScutari, Rossana, Valentino Costabile, Laura Galli, Maria Concetta Bellocchi, Luca Carioti, Silvia Barbaliscia, Andrea Poli, Andrea Galli, Carlo Federico Perno, Maria Mercedes Santoro, and et al. 2021. "Impact of Analytical Treatment Interruption on Burden and Diversification of HIV Peripheral Reservoir: A Pilot Study" Viruses 13, no. 7: 1403. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071403