
Structure-Guided Creation of an Anti-HA Stalk Antibody F11 Derivative That Neutralizes Both F11-Sensitive and -Resistant Influenza A(H1N1)pdm09 Viruses

 , ,
, , 
{kind=link}
{kind=link}
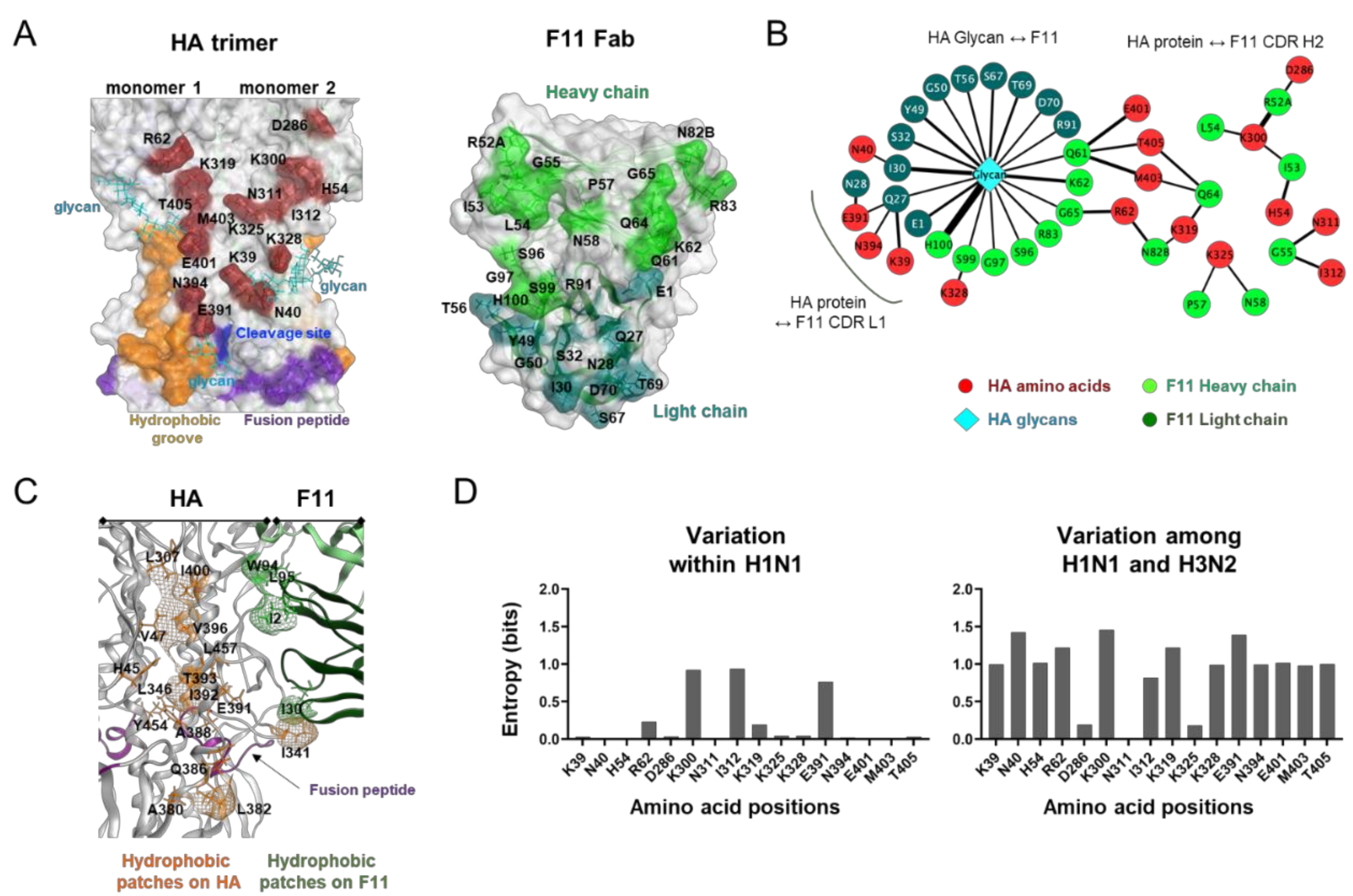{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Modeling of the Trimeric HA Ectodomain of Influenza A(H1N1)pdm09
2.2. Docking Simulations of the Glycosylated HA Ectodomain and Anti-HA Stalk Antibody F11
2.3. MD Simulation of the Glycosylated HA Trimer Docked to the F11 Fab Fragment
2.4. Calculation of Root Mean Square Deviation (RMSD)
2.5. Calculation of Root Mean Square Fluctuation (RMSF)
2.6. Analysis of Noncovalent Interaction Sites and Networks between HA and F11
2.7. Protein Patch Analysis
2.8. Shannon Entropy Analysis
2.9. In Silico Site-Directed Mutagenesis of the F11 Antibody
2.10. Cells and Viruses
2.11. Site-Directed Mutagenesis, Expression, and Purification of IgG Antibodies
2.12. Assessment of Mutated IgG1 Antibody Quality by SDS-PAGE Analysis
2.13. Microneutralization (NT) Assay
2.14. Statistical Analysis
3. Results
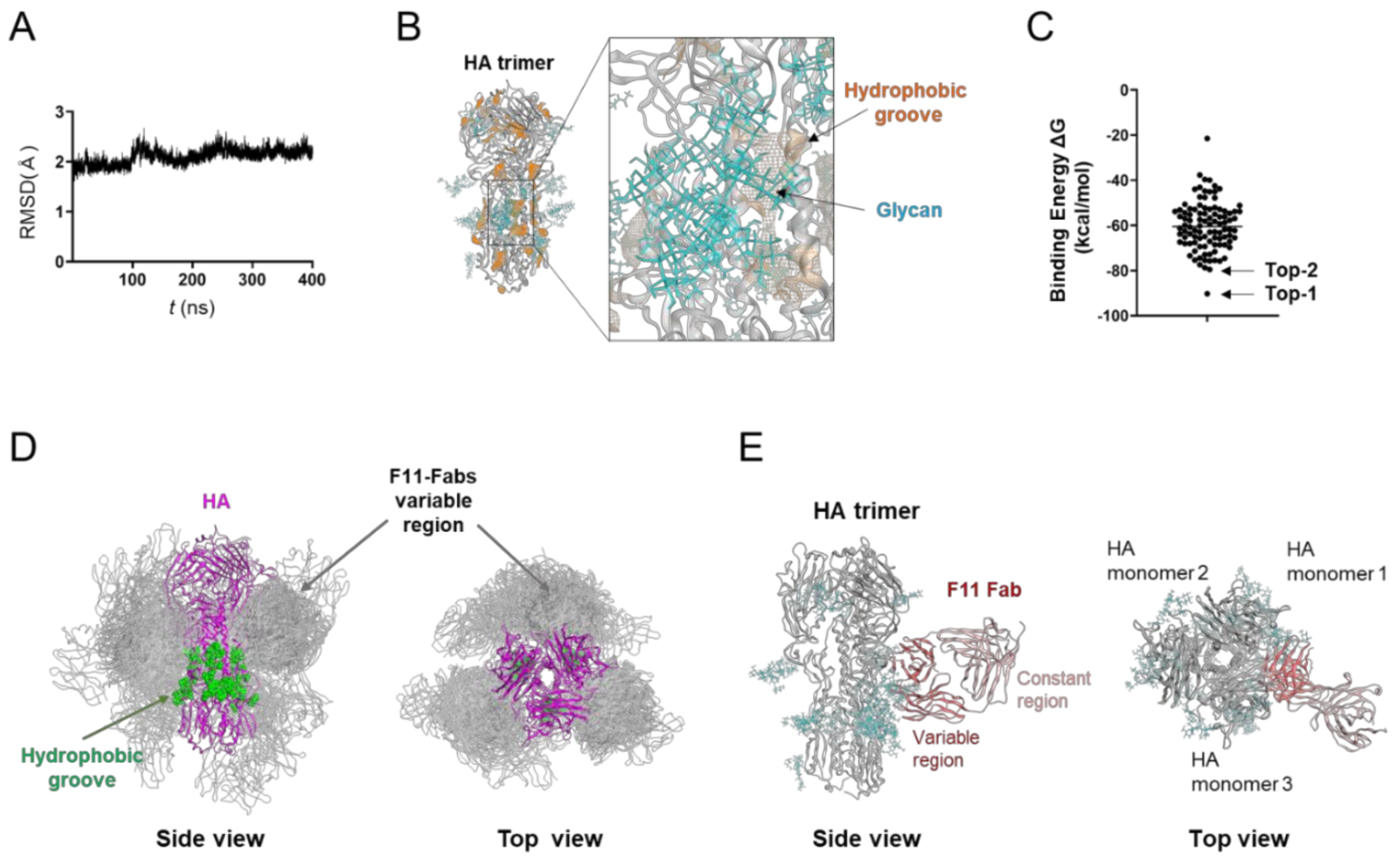
3.1. Molecular Modeling of the Glycosylated HA Trimer Ectodomain Docked to the F11 Fab Fragment
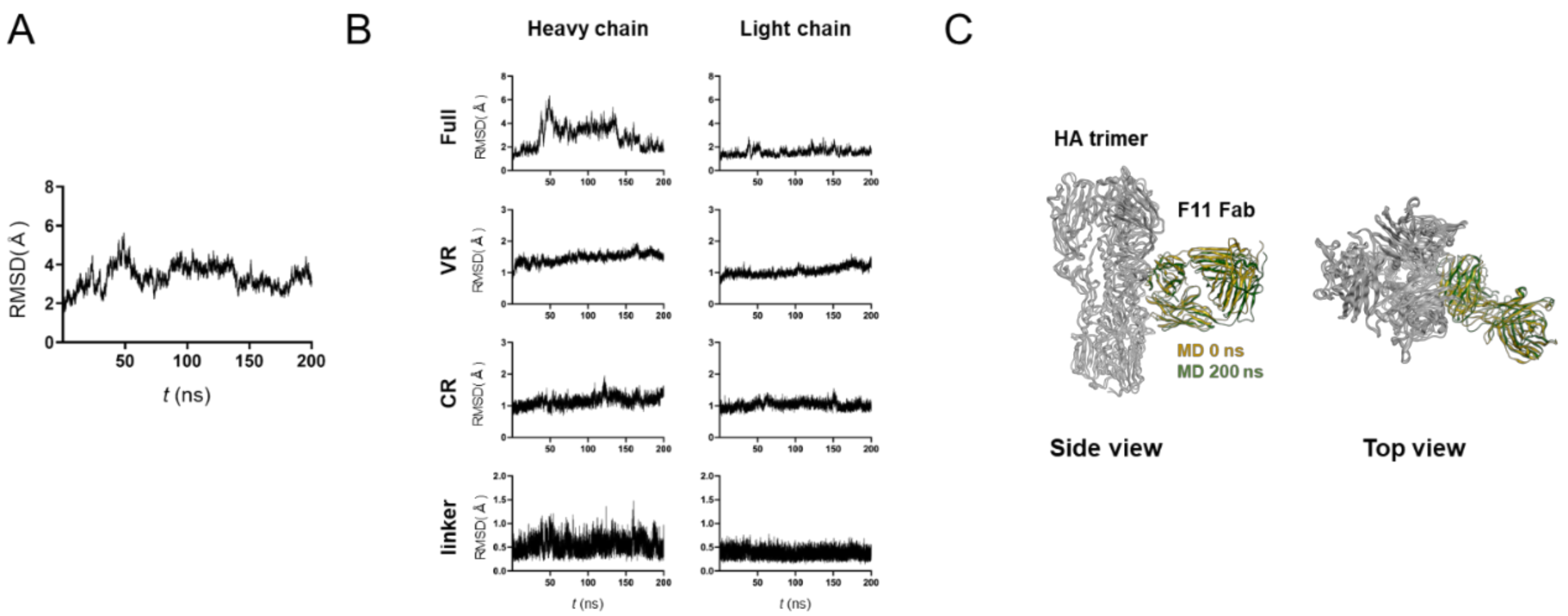
3.2. MD Simulation of Glycosylated HA Trimer Docked to F11 Fab Fragment
3.3. Characterization of Molecular Interactions between the HA Trimer Ectodomain and F11 Fab Fragment
3.4. Effects of F11 Fab Binding on the Structural Fluctuations of the HA Ectodomain
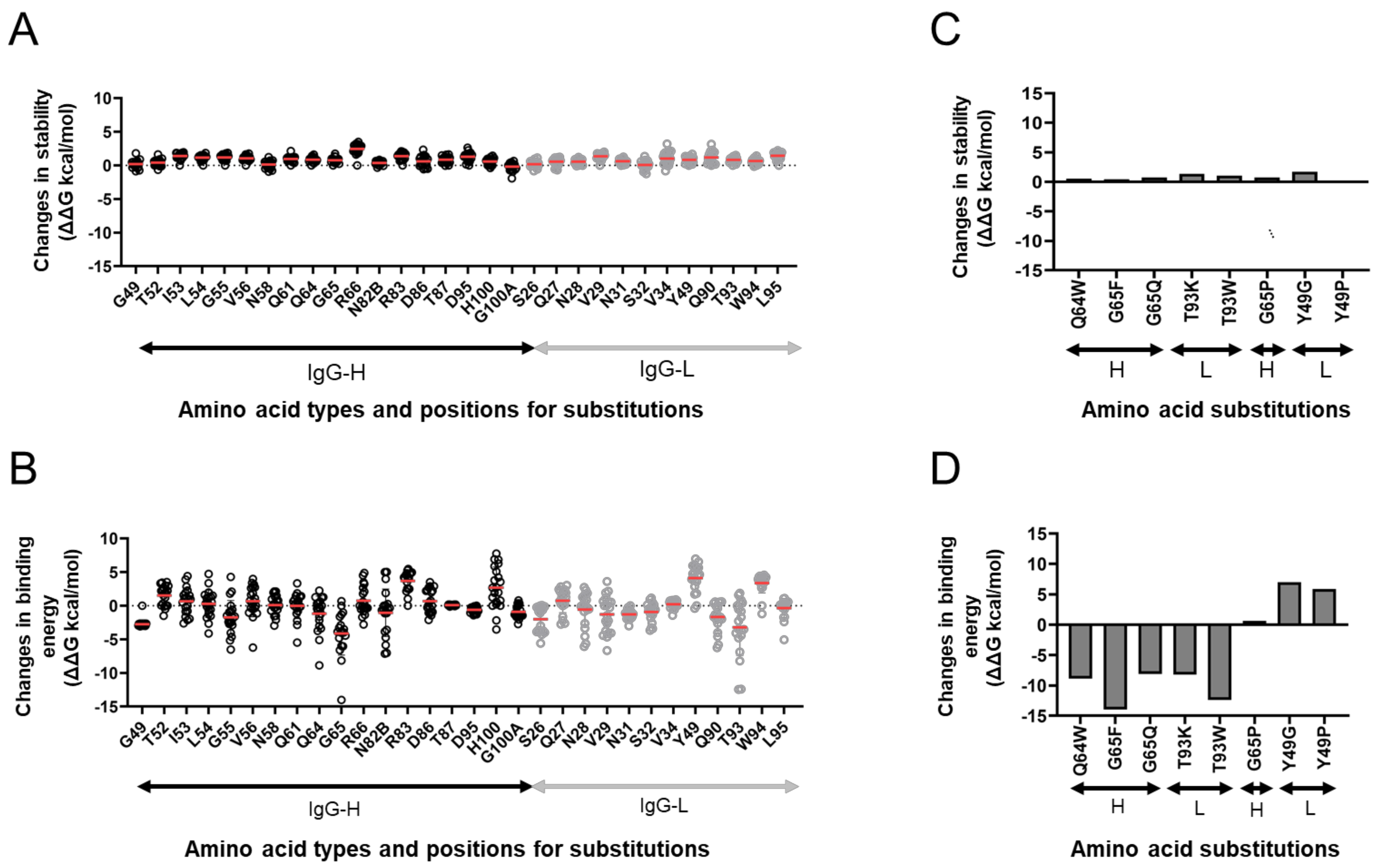
3.5. In Silico Site-Directed Mutagenesis
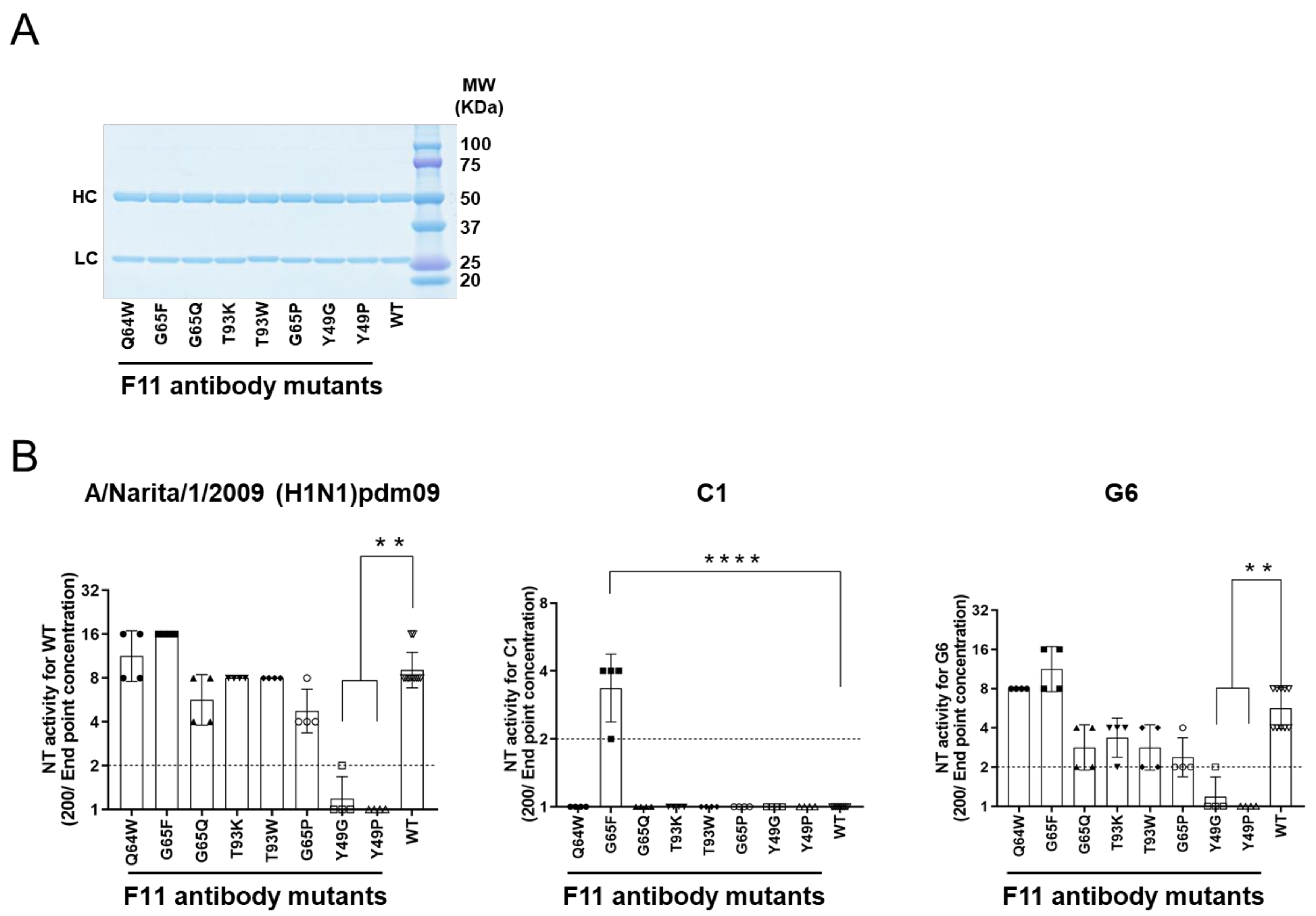
3.6. Experimental Mutagenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CDR | complementarity determining region |
| CR | constant region |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| EHT | Extended Huckel Theory |
| HA | Hemagglutinin |
| hCK | Humanized MDCK |
| MD | Molecular dynamics |
| MDCK | Madin-Darby canine kidney |
| MEM | Minimum essential medium |
| MOE | Molecular Operating Environment |
| NT | microneutralization |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| VR | variable region |
References
- Altman, M.O.; Angeletti, D.; Yewdell, J.W. Antibody immunodominance: The key to understanding influenza virus antigenic drift. Viral Immunol. 2018, 31, 142–149. [Google Scholar] [CrossRef]
- Wu, N.C.; Wilson, I.A. A perspective on the structural and functional constraints for immune evasion: Insights from influenza virus. J. Mol. Biol. 2017, 429, 2694–2709. [Google Scholar] [CrossRef]
- Bhatt, S.; Holmes, E.C.; Pybus, O.G. The genomic rate of molecular adaptation of the human influenza a virus. Mol. Biol. Evol. 2011, 28, 2443–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkpatrick, E.; Qiu, X.; Wilson, P.C.; Bahl, J.; Krammer, F. The influenza virus hemagglutinin head evolves faster than the stalk domain. Sci. Rep. 2018, 8, 10432. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza a viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Palese, P. Overcoming barriers in the path to a universal influenza virus vaccine. Cell Host Microbe 2018, 24, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadlbauer, D.; Nachbagauer, R.; Meade, P.; Krammer, F. Universal influenza virus vaccines: What can we learn from the human immune response following exposure to H7 subtype viruses? Front. Med. 2017, 11, 471–479. [Google Scholar] [CrossRef]
- Sedeyn, K.; Saelens, X. New antibody-based prevention and treatment options for influenza. Antivir. Res. 2019, 170, 104562. [Google Scholar] [CrossRef]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef]
- Roubidoux, E.K.; Carreno, J.M.; McMahon, M.; Jiang, K.; van Bakel, H.; Wilson, P.; Krammer, F. Mutations in the hemagglutinin stalk domain do not permit escape from a protective, stalk-based vaccine-induced immune response in the mouse model. mBio 2021, 12, e03617-20. [Google Scholar] [CrossRef]
- Benton, D.J.; Gamblin, S.J.; Rosenthal, P.B.; Skehel, J.J. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature 2020, 583, 150–153. [Google Scholar] [CrossRef]
- Joyce, M.G.; Wheatley, A.K.; Thomas, P.V.; Chuang, G.Y.; Soto, C.; Bailer, R.T.; Druz, A.; Georgiev, I.S.; Gillespie, R.A.; Kanekiyo, M.; et al. Vaccine-Induced antibodies that neutralize group 1 and group 2 influenza a viruses. Cell 2016, 166, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Cho, M.; Shore, D.; Song, M.; Choi, J.; Jiang, T.; Deng, Y.Q.; Bourgeois, M.; Almli, L.; Yang, H.; et al. A potent broad-spectrum protective human monoclonal antibody crosslinking two haemagglutinin monomers of influenza A virus. Nat. Commun. 2015, 6, 7708. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.A.; Friesen, R.H.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody recognition of a highly conserved influenza virus epitope. Science 2009, 324, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Dreyfus, C.; Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongeneelen, M.; et al. Highly conserved protective epitopes on influenza b viruses. Science 2012, 337, 1343–1348. [Google Scholar] [CrossRef] [Green Version]
- Kallewaard, N.L.; Corti, D.; Collins, P.J.; Neu, U.; McAuliffe, J.M.; Benjamin, E.; Wachter-Rosati, L.; Palmer-Hill, F.J.; Yuan, A.Q.; Walker, P.A.; et al. Structure and function analysis of an antibody recognizing all influenza a subtypes. Cell 2016, 166, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Xie, J.; Zhu, X.; Wu, N.C.; Lerner, R.A.; Wilson, I.A. Antibody 27F3 broadly targets influenza a group 1 and 2 hemagglutinins through a further variation in VH1-69 antibody orientation on the HA stem. Cell Rep. 2017, 20, 2935–2943. [Google Scholar] [CrossRef] [Green Version]
- Sano, K.; Saito, S.; Suzuki, T.; Kotani, O.; Ainai, A.; van Riet, E.; Tabata, K.; Saito, K.; Takahashi, Y.; Yokoyama, M.; et al. An influenza HA stalk reactive polymeric IgA antibody exhibits anti-viral function regulated by binary interaction between HA and the antibody. PLoS ONE 2021, 16, e0245244. [Google Scholar] [CrossRef]
- Ainai, A.; Hasegawa, H.; Obuchi, M.; Odagiri, T.; Ujike, M.; Shirakura, M.; Nobusawa, E.; Tashiro, M.; Asanuma, H. Host adaptation and the alteration of viral properties of the first influenza A/H1N1pdm09 virus isolated in Japan. PLoS ONE 2015, 10, e0130208. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Ekiert, D.C.; Krause, J.C.; Hai, R.; Crowe, J.E., Jr.; Wilson, I.A. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010, 328, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GLYCAM-Web. Available online: http://glycam.org (accessed on 1 March 2018).
- Yokoyama, M.; Fujisaki, S.; Shirakura, M.; Watanabe, S.; Odagiri, T.; Ito, K.; Sato, H. Molecular dynamics simulation of the influenza A(H3N2) hemagglutinin trimer reveals the structural basis for adaptive evolution of the recent epidemic clade 3C.2a. Front. Microbiol. 2017, 8, 584. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; Gonzalez-Outeirino, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impei, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.K.; Labute, P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 2014, 82, 1599–1610. [Google Scholar] [CrossRef] [Green Version]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput.-Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koma, T.; Kotani, O.; Miyakawa, K.; Ryo, A.; Yokoyama, M.; Doi, N.; Adachi, A.; Sato, H.; Nomaguchi, M. Allosteric regulation of HIV-1 capsid structure for gag assembly, virion production, and viral infectivity by a disordered interdomain linker. J. Virol. 2019, 93, e00381-19. [Google Scholar] [CrossRef] [Green Version]
- Koma, T.; Yokoyama, M.; Kotani, O.; Doi, N.; Nakanishi, N.; Okubo, H.; Adachi, S.; Adachi, A.; Sato, H.; Nomaguchi, M. Species-Specific Valid ternary interactions of HIV-1 Env-gp120, CD4, and CCR5 as revealed by an adaptive single-amino acid substitution at the V3 loop tip. J. Virol. 2021, 95, e0217720. [Google Scholar] [CrossRef]
- Sakuragi, S.; Kotani, O.; Yokoyama, M.; Shioda, T.; Sato, H.; Sakuragi, J.I. Identification of a novel cis-acting regulator of HIV-1 genome packaging. Int. J. Mol. Sci. 2021, 22, 3435. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Capponi, S.; Geuens, T.; Geroldi, A.; Origone, P.; Verdiani, S.; Cichero, E.; Adriaenssens, E.; De Winter, V.; Bandettini di Poggio, M.; Barberis, M.; et al. Molecular chaperones in the pathogenesis of amyotrophic lateral sclerosis: The role of HSPB1. Hum. Mutat. 2016, 37, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Dindo, M.; Montioli, R.; Busato, M.; Giorgetti, A.; Cellini, B.; Borri Voltattorni, C. Effects of interface mutations on the dimerization of alanine glyoxylate aminotransferase and implications in the mistargeting of the pathogenic variants F152I and I244T. Biochimie 2016, 131, 137–148. [Google Scholar] [CrossRef]
- Jetha, A.; Thorsteinson, N.; Jmeian, Y.; Jeganathan, A.; Giblin, P.; Fransson, J. Homology modeling and structure-based design improve hydrophobic interaction chromatography behavior of integrin binding antibodies. MAbs 2018, 10, 890–900. [Google Scholar] [CrossRef]
- Shah, M.; Anwar, M.A.; Park, S.; Jafri, S.S.; Choi, S. In silico mechanistic analysis of IRF3 inactivation and high-risk HPV E6 species-dependent drug response. Sci. Rep. 2015, 5, 13446. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.J.; Kumar, S.; Wang, X.; Helk, B.; Singh, S.K.; Trout, B.L. Aggregation in protein-based biotherapeutics: Computational studies and tools to identify aggregation-prone regions. J. Pharm. Sci. 2011, 100, 5081–5095. [Google Scholar] [CrossRef]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [Green Version]
- Motomura, K.; Oka, T.; Yokoyama, M.; Nakamura, H.; Mori, H.; Ode, H.; Hansman, G.S.; Katayama, K.; Kanda, T.; Tanaka, T.; et al. Identification of monomorphic and divergent haplotypes in the 2006-2007 norovirus GII/4 epidemic population by genomewide tracing of evolutionary history. J. Virol. 2008, 82, 11247–11262. [Google Scholar] [CrossRef] [Green Version]
- GISAID. Available online: https://www.gisaid.org. (accessed on 1 June 2018).
- Shannon, C.E. The mathematical theory of communication. 1963. MD Comput. 1997, 14, 306–317. [Google Scholar]
- Takashita, E.; Fujisaki, S.; Yokoyama, M.; Shirakura, M.; Morita, H.; Nakamura, K.; Kishida, N.; Kuwahara, T.; Sato, H.; Doi, I.; et al. In Vitro characterization of multidrug-resistant influenza A(H1N1)pdm09 viruses carrying a dual neuraminidase mutation isolated from immunocompromised patients. Pathogens 2020, 9, 725. [Google Scholar] [CrossRef]
- Takashita, E.; Kiso, M.; Fujisaki, S.; Yokoyama, M.; Nakamura, K.; Shirakura, M.; Sato, H.; Odagiri, T.; Kawaoka, Y.; Tashiro, M. Characterization of a large cluster of influenza A(H1N1)pdm09 viruses cross-resistant to oseltamivir and peramivir during the 2013-2014 influenza season in Japan. Antimicrob. Agents Chemother. 2015, 59, 2607–2617. [Google Scholar] [CrossRef] [Green Version]
- Nomaguchi, M.; Yokoyama, M.; Kono, K.; Nakayama, E.E.; Shioda, T.; Doi, N.; Fujiwara, S.; Saito, A.; Akari, H.; Miyakawa, K.; et al. Generation of rhesus macaque-tropic HIV-1 clones that are resistant to major anti-HIV-1 restriction factors. J. Virol. 2013, 87, 11447–11461. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Kawakami, C.; Fan, S.; Chiba, S.; Zhong, G.; Gu, C.; Shimizu, K.; Takasaki, S.; Sakai-Tagawa, Y.; Lopes, T.J.S.; et al. A humanized MDCK cell line for the efficient isolation and propagation of human influenza viruses. Nat. Microbiol. 2019, 4, 1268–1273. [Google Scholar] [CrossRef]
- Ainai, A.; Tamura, S.; Suzuki, T.; van Riet, E.; Ito, R.; Odagiri, T.; Tashiro, M.; Kurata, T.; Hasegawa, H. Intranasal vaccination with an inactivated whole influenza virus vaccine induces strong antibody responses in serum and nasal mucus of healthy adults. Hum. Vaccin. Immunother. 2013, 9, 1962–1970. [Google Scholar] [CrossRef]
- Saito, S.; Sano, K.; Suzuki, T.; Ainai, A.; Taga, Y.; Ueno, T.; Tabata, K.; Saito, K.; Wada, Y.; Ohara, Y.; et al. IgA tetramerization improves target breadth but not peak potency of functionality of anti-influenza virus broadly neutralizing antibody. PLoS Pathog. 2019, 15, e1007427. [Google Scholar] [CrossRef] [Green Version]
- Knipe, D.M.; Howley, P.M. Orthomyxoviridae. Fields Virol. 2013, 1, 1151–1185. [Google Scholar]
- Harshbarger, W.D.; Deming, D.; Lockbaum, G.J.; Attatippaholkun, N.; Kamkaew, M.; Hou, S.; Somasundaran, M.; Wang, J.P.; Finberg, R.W.; Zhu, Q.K.; et al. Unique structural solution from a VH3-30 antibody targeting the hemagglutinin stem of influenza a viruses. Nat. Commun. 2021, 12, 559. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Yamayoshi, S.; Ito, M.; Uraki, R.; Kawaoka, Y.; Wilson, I.A. Recurring and adaptable binding motifs in broadly neutralizing antibodies to influenza virus are encoded on the D3-9 segment of the ig gene. Cell Host Microbe 2018, 24, 569–578.e4. [Google Scholar] [CrossRef] [Green Version]
- Dreyfus, C.; Ekiert, D.C.; Wilson, I.A. Structure of a classical broadly neutralizing stem antibody in complex with a pandemic H2 influenza virus hemagglutinin. J. Virol. 2013, 87, 7149–7154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath Neerukonda, S.; Vassell, R.; Weiss, C.D. Neutralizing antibodies targeting the conserved stem region of influenza hemagglutinin. Vaccines 2020, 8, 382. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Kath, J.E.; Zorgiebel, F.M.; Sun, M.; Ghosh, P.; Hatfull, G.F.; Grindley, N.D.; Marko, J.F. Remote control of DNA-acting enzymes by varying the brownian dynamics of a distant DNA end. Proc. Natl. Acad. Sci. USA 2012, 109, 16546–16551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, G.G.; Lane, D.P.; Verma, C.S. Molecular simulations of protein dynamics: New windows on mechanisms in biology. EMBO Rep. 2008, 9, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA 2005, 102, 6679–6685. [Google Scholar] [CrossRef] [Green Version]
- Keul, N.D.; Oruganty, K.; Schaper Bergman, E.T.; Beattie, N.R.; McDonald, W.E.; Kadirvelraj, R.; Gross, M.L.; Phillips, R.S.; Harvey, S.C.; Wood, Z.A. The entropic force generated by intrinsically disordered segments tunes protein function. Nature 2018, 563, 584–588. [Google Scholar] [CrossRef]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef] [Green Version]
- Callan, R.J.; Hartmann, F.A.; West, S.E.; Hinshaw, V.S. Cleavage of influenza a virus H1 hemagglutinin by swine respiratory bacterial proteases. J. Virol. 1997, 71, 7579–7585. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, B.S.; Whittaker, G.R.; Daniel, S. Influenza virus-mediated membrane fusion: Determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses 2012, 4, 1144–1168. [Google Scholar] [CrossRef] [Green Version]
- Stieneke-Grober, A.; Vey, M.; Angliker, H.; Shaw, E.; Thomas, G.; Roberts, C.; Klenk, H.D.; Garten, W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992, 11, 2407–2414. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Shi, Y.; Lu, X.; He, J.; Gao, F.; Yan, J.; Qi, J.; Gao, G.F. Bat-Derived influenza hemagglutinin H17 does not bind canonical avian or human receptors and most likely uses a unique entry mechanism. Cell Rep. 2013, 3, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 226–249. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Peng, W.; Qi, J.; Chai, Y.; Song, H.; Bi, Y.; Rijal, P.; Wang, H.; Oladejo, B.O.; Liu, J.; et al. Structure-Based modification of an anti-neuraminidase human antibody restores protection efficacy against the drifted influenza virus. mBio 2020, 11, e02315-20. [Google Scholar] [CrossRef] [PubMed]
- Bottcher-Friebertshauser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef] [Green Version]
- Dou, D.; Revol, R.; Ostbye, H.; Wang, H.; Daniels, R. Influenza a virus cell entry, replication, virion assembly and movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Liu, W.C.; Jan, J.T.; Huang, Y.J.; Chen, T.H.; Wu, S.C. Unmasking stem-specific neutralizing epitopes by abolishing n-linked glycosylation sites of influenza virus hemagglutinin proteins for vaccine design. J. Virol. 2016, 90, 8496–8508. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotani, O.; Suzuki, Y.; Saito, S.; Ainai, A.; Ueno, A.; Hemmi, T.; Sano, K.; Tabata, K.; Yokoyama, M.; Suzuki, T.; et al. Structure-Guided Creation of an Anti-HA Stalk Antibody F11 Derivative That Neutralizes Both F11-Sensitive and -Resistant Influenza A(H1N1)pdm09 Viruses. Viruses 2021, 13, 1733. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091733
Kotani O, Suzuki Y, Saito S, Ainai A, Ueno A, Hemmi T, Sano K, Tabata K, Yokoyama M, Suzuki T, et al. Structure-Guided Creation of an Anti-HA Stalk Antibody F11 Derivative That Neutralizes Both F11-Sensitive and -Resistant Influenza A(H1N1)pdm09 Viruses. Viruses. 2021; 13(9):1733. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091733
Chicago/Turabian StyleKotani, Osamu, Yasushi Suzuki, Shinji Saito, Akira Ainai, Akira Ueno, Takuya Hemmi, Kaori Sano, Koshiro Tabata, Masaru Yokoyama, Tadaki Suzuki, and et al. 2021. "Structure-Guided Creation of an Anti-HA Stalk Antibody F11 Derivative That Neutralizes Both F11-Sensitive and -Resistant Influenza A(H1N1)pdm09 Viruses" Viruses 13, no. 9: 1733. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091733