
Mouse Papillomavirus L1 and L2 Are Dispensable for Viral Infection and Persistence at Both Cutaneous and Mucosal Tissues

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
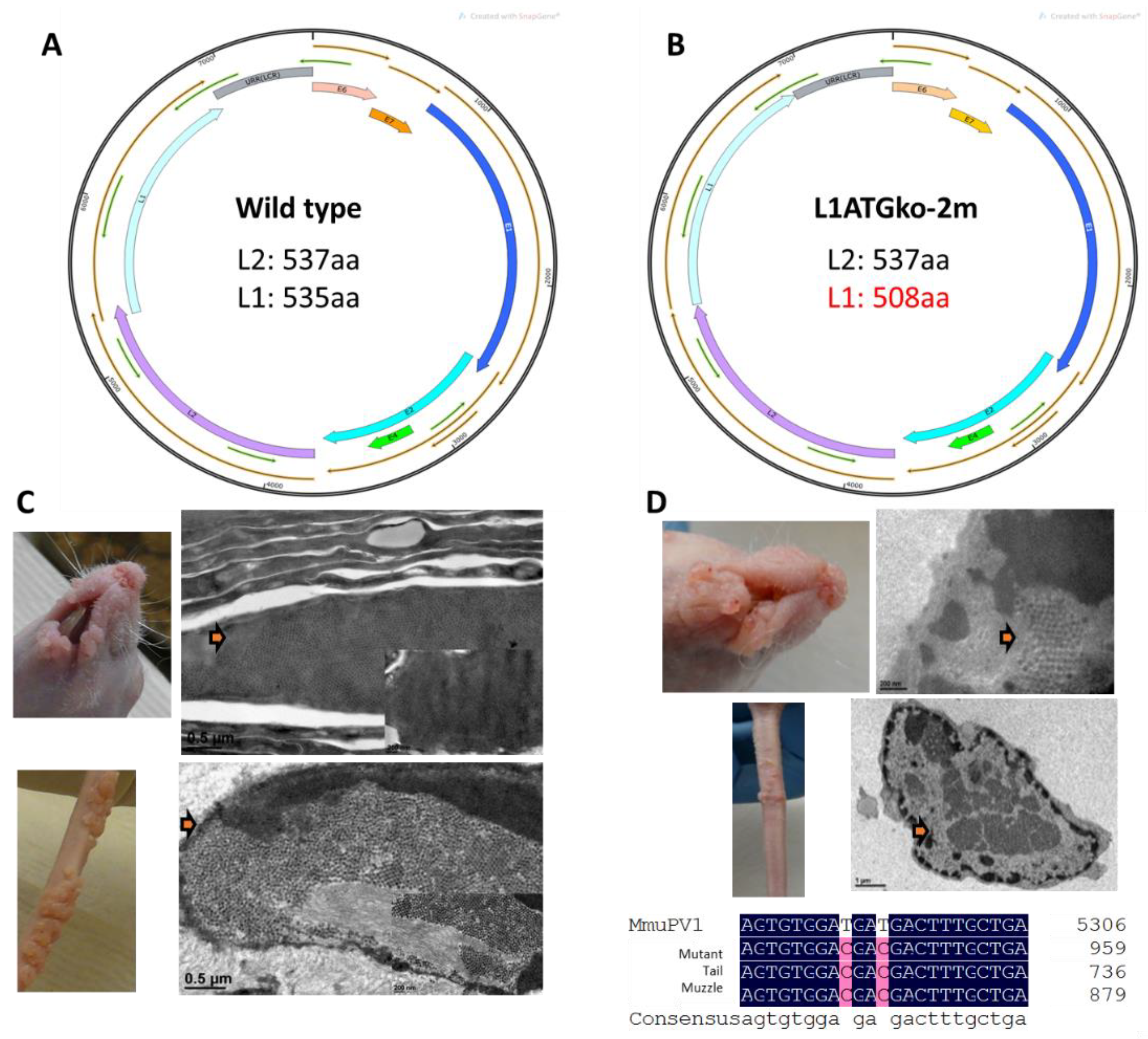
2.1. Mutant Genome Generation
2.2. Animals and Viral Infection
2.3. Detection of Viral DNA by qPCR
2.4. Confirmation of Viral DNA by Rolling Circle Amplification and Sequencing
2.5. Detection of Virus by ELISA, In Situ Hybridization, Immunohistochemistry, and Transmission Electron Microscope
2.6. Western Blot Analysis for Detection of L1 Protein
2.7. Statistical Analysis
3. Results
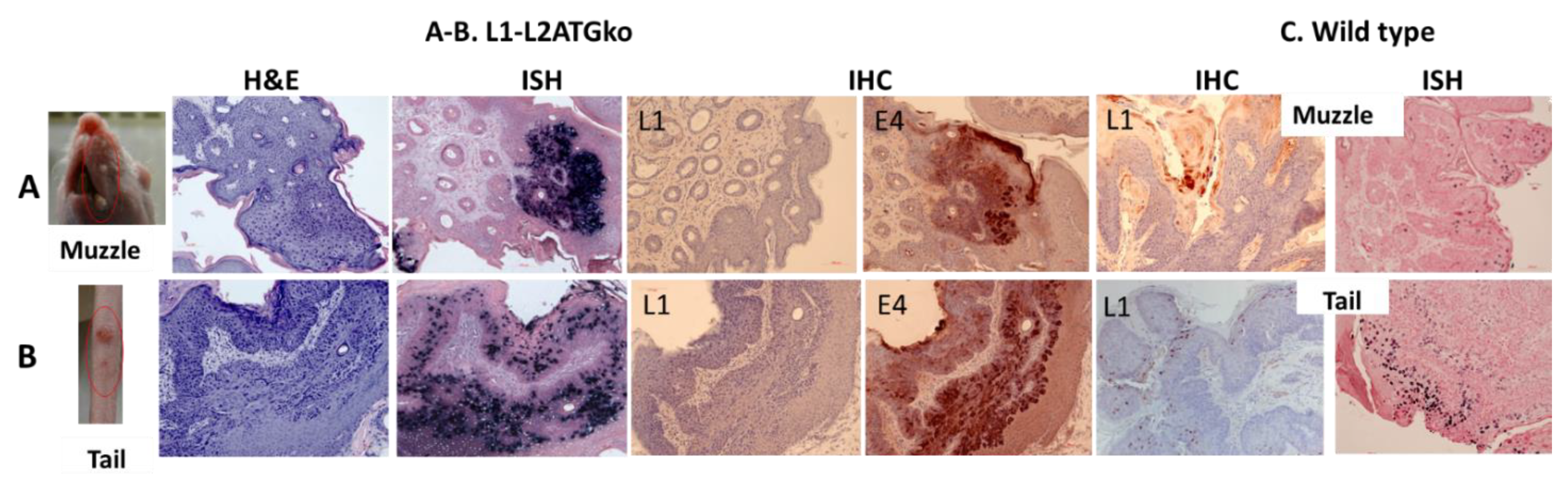
3.1. Persistent Lesions Were Induced by L1ATGko-2m at Cutaneous Sites
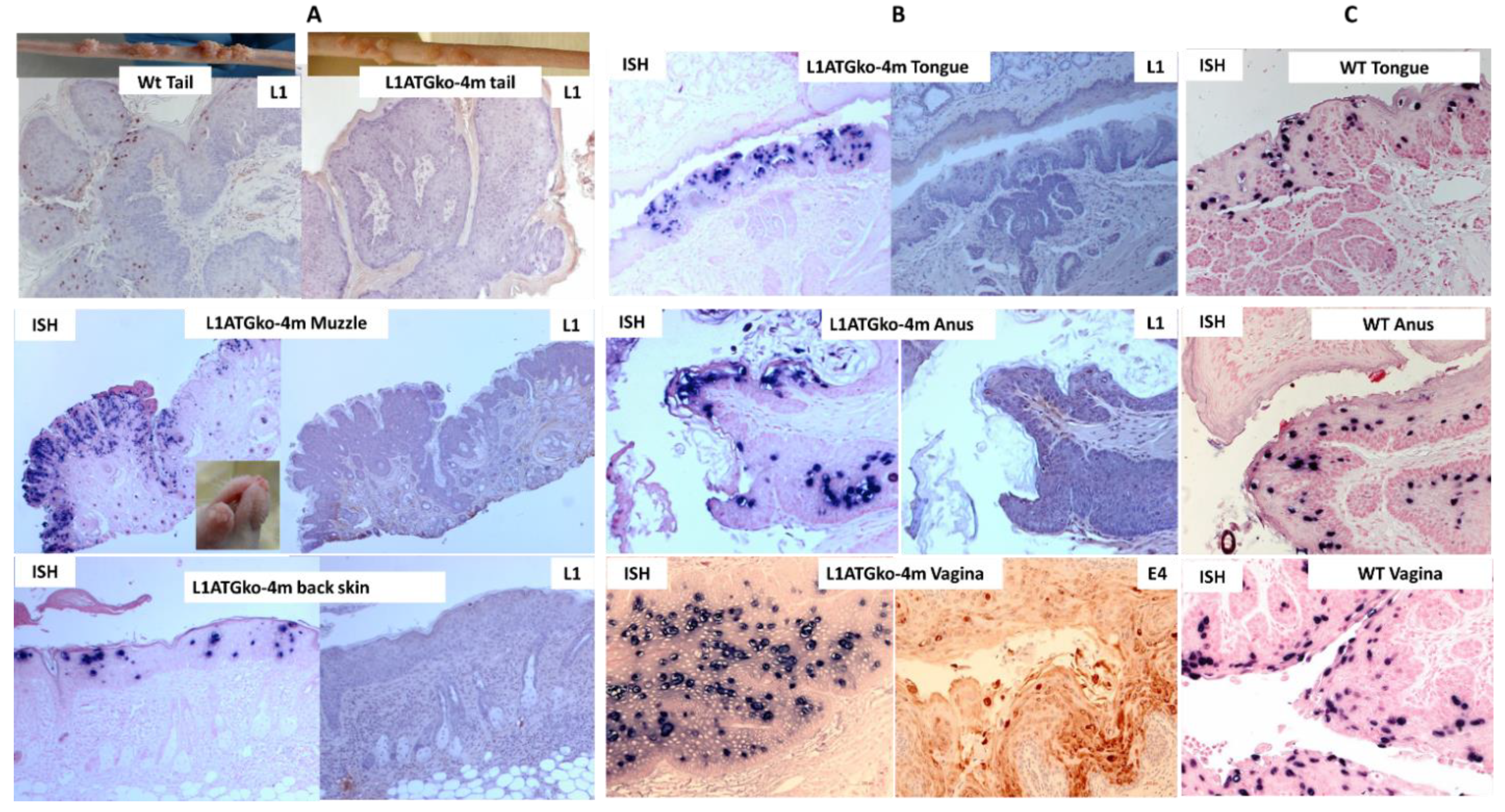
3.2. The L1ATGko-4m Mutant Induced Persistent Infections at Cutaneous and Mucosal Sites
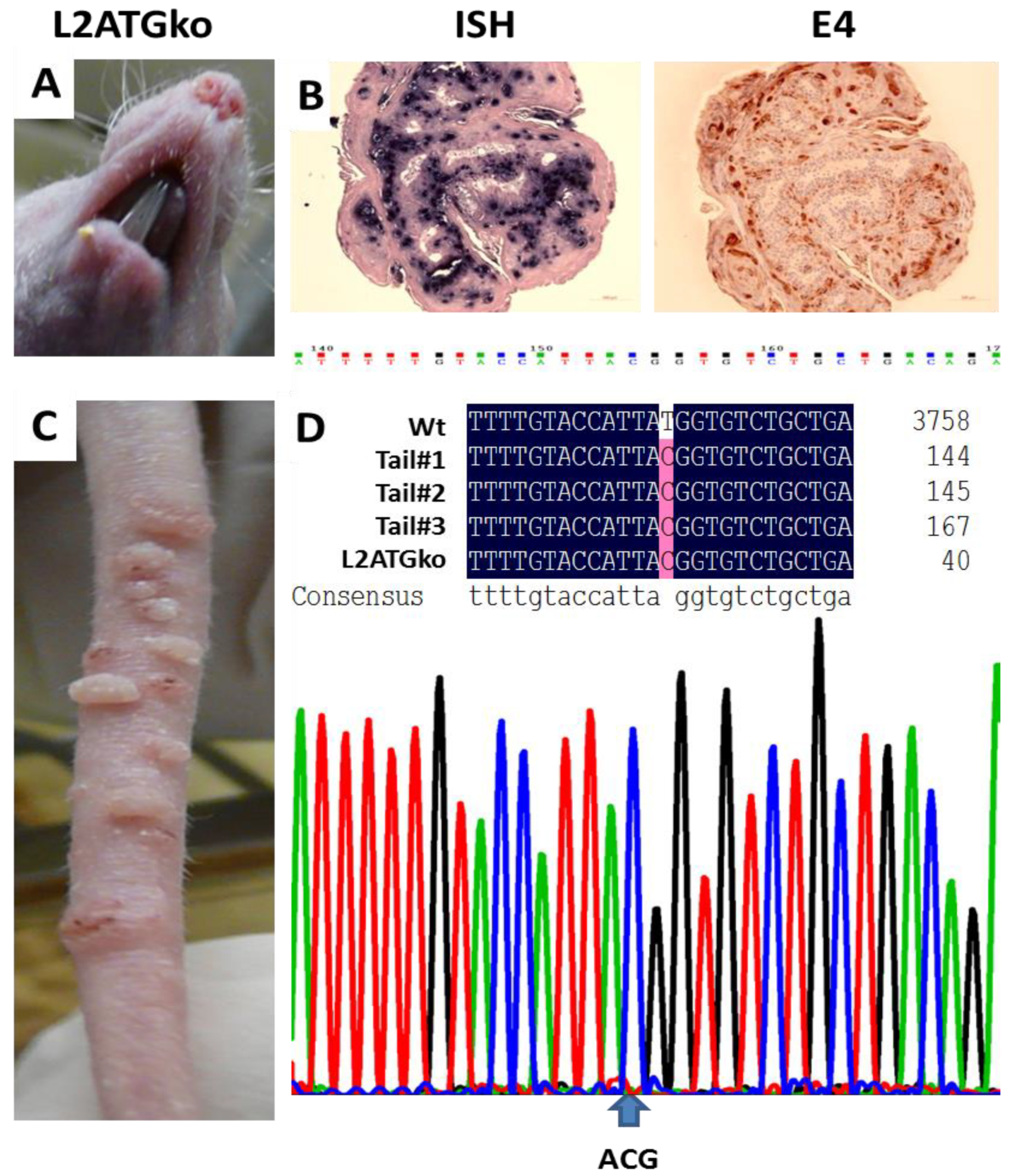
3.3. L2 Is Not Required for Tumor Growth in Cutaneous Tissues of Nude Mice
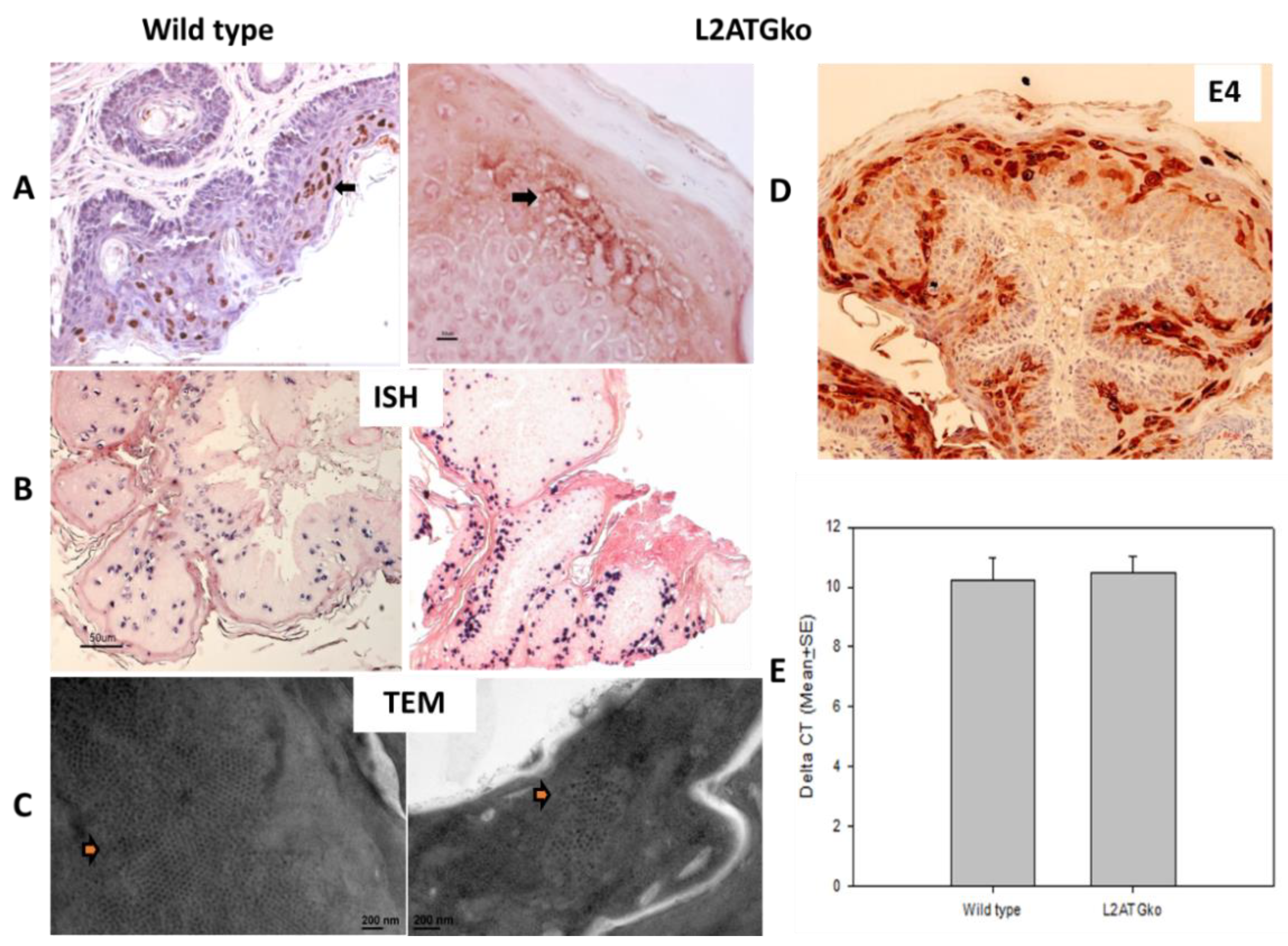
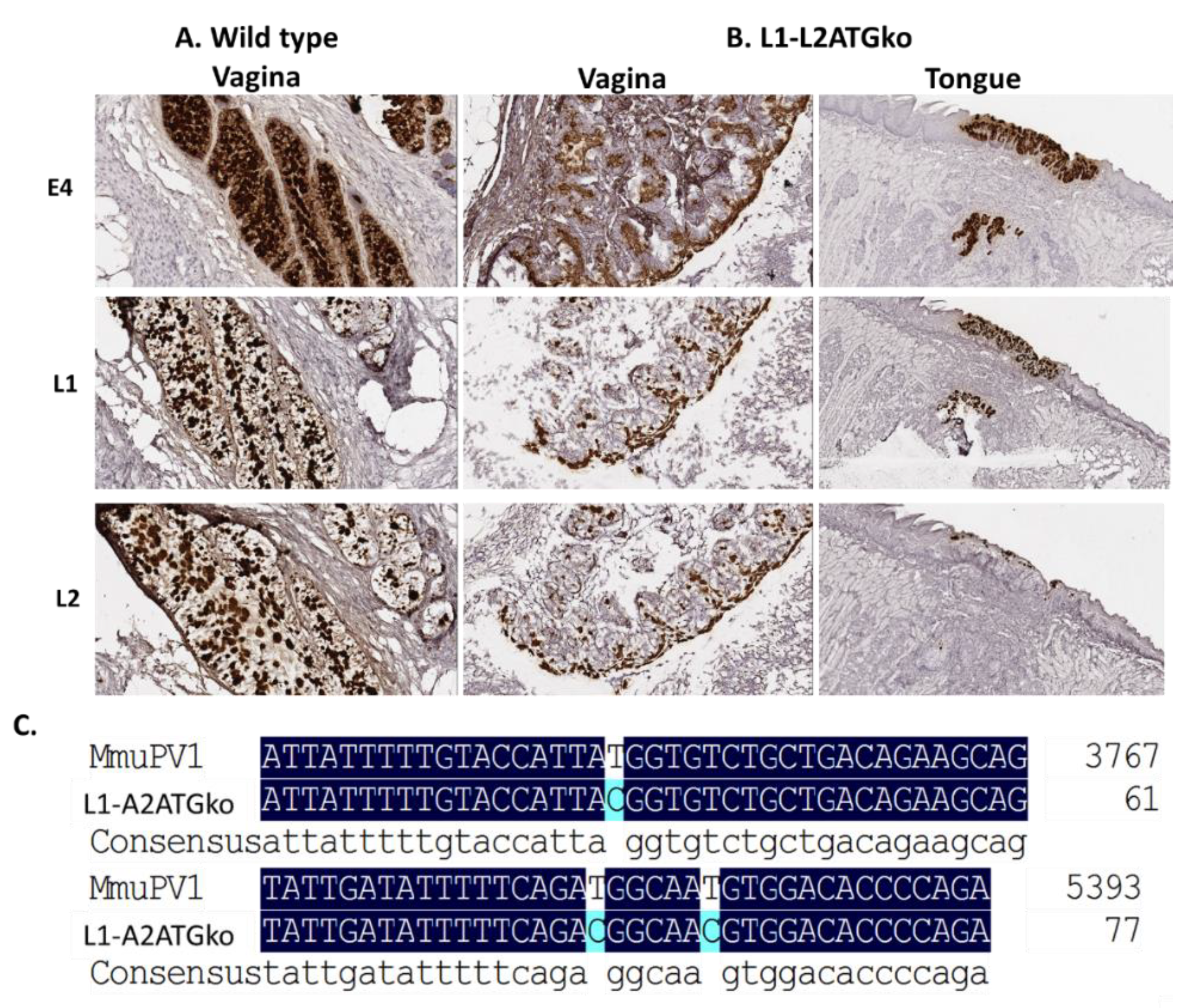
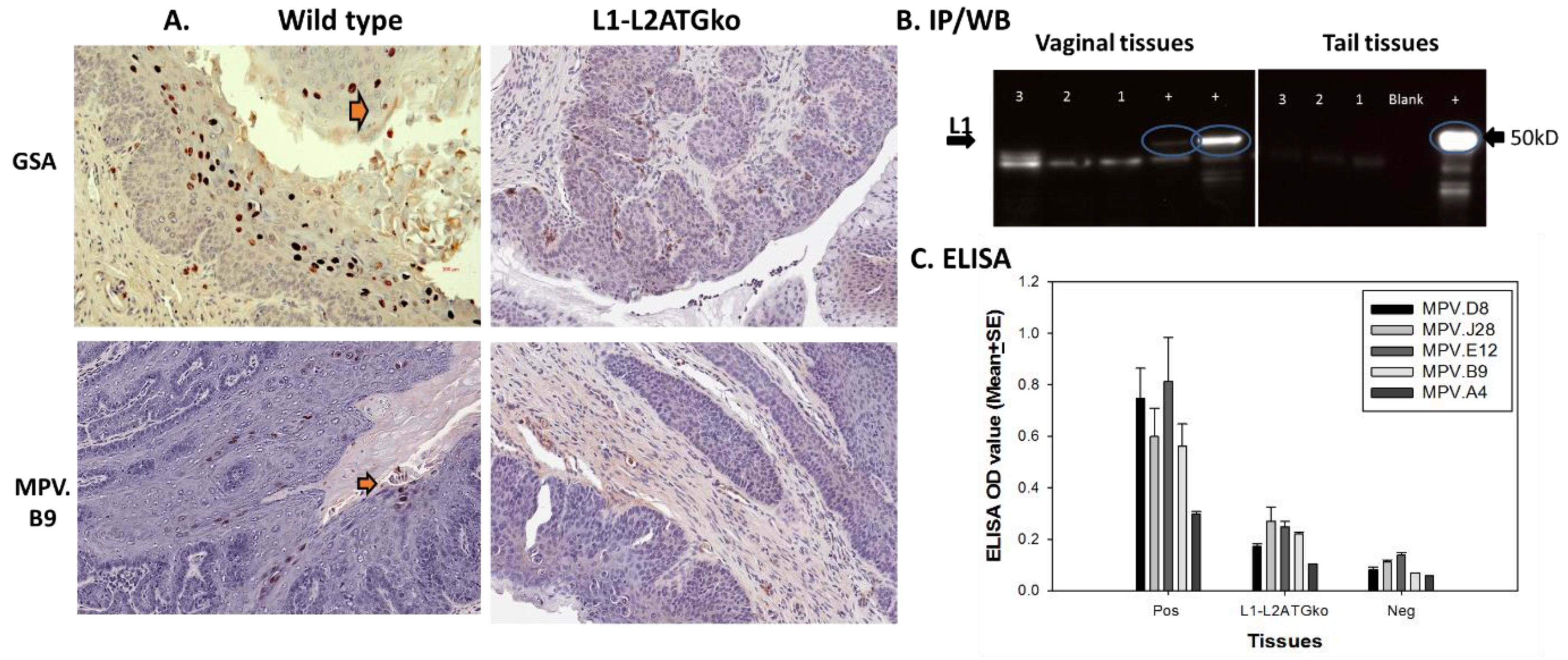
3.4. L2 Is Not Required for L1 Expression but Influences Full-Length L1 Location in Infected Cells
3.5. Simultaneous Mutations of L1 and L2 Do Not Influence Viral DNA Amplification
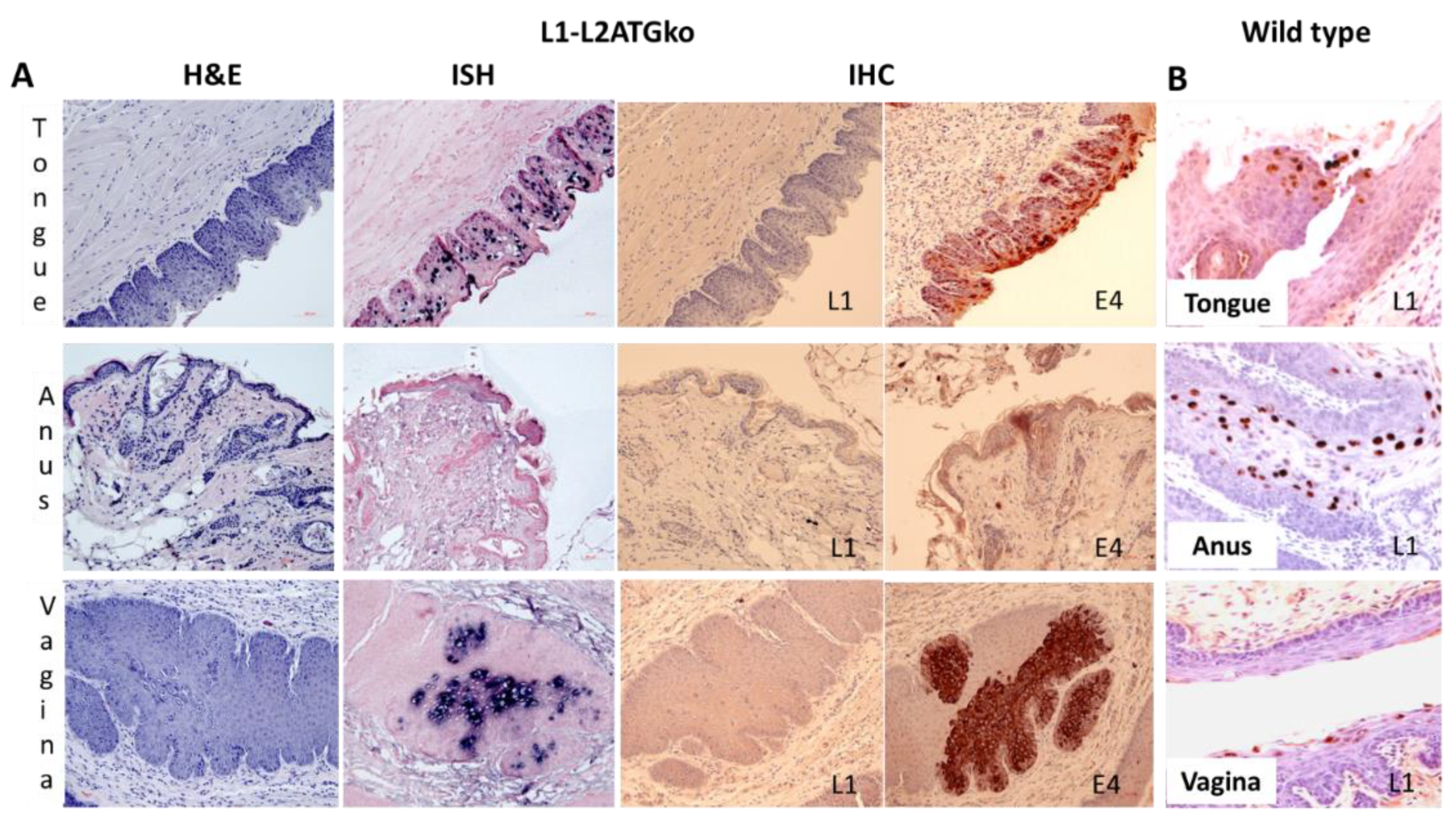
3.6. Differential Infection Patterns Were Noted in Mucosal Infections with L1 and L2ATGko Mutants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MmuPV1 | the mouse papillomavirus |
| IHC | immunohistochemistry |
| ISH | in situ hybridization |
| RNA-ISH | RNA-in situ hybridization |
| IP | immunoprecipitation |
| TEM | Transmission electron microscopy |
References
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L1. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Christensen, N.D.; Budgeon, L.R.; Cladel, N.M.; Hu, J. Recent advances in preclinical model systems for papillomaviruses. Virus Res. 2017, 231, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Nasseri, M.; Meyers, C.; Wettstein, F.O. Genetic analysis of CRPV pathogenesis: The L1 open reading frame is dispensable for cellular transformation but is required for papilloma formation. Virology 1989, 170, 321–325. [Google Scholar] [CrossRef]
- Cladel, N.M.; Peng, X.; Christensen, N.; Hu, J. The rabbit papillomavirus model: A valuable tool to study viral-host interactions. Philos. Trans. R Soc. Lond. B Biol. Sci. 2019, 374, 20180294. [Google Scholar] [CrossRef] [Green Version]
- Embers, M.E.; Budgeon, L.R.; Pickel, M.; Christensen, N.D. Protective immunity to rabbit oral and cutaneous papillomaviruses by immunization with short peptides of l2, the minor capsid protein. J. Virol. 2002, 76, 9798–9805. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Budgeon, L.R.; Cladel, N.M.; Culp, T.D.; Balogh, K.K.; Christensen, N.D. Detection of L1, infectious virions and anti-L1 antibody in domestic rabbits infected with cottontail rabbit papillomavirus. J. Gen. Virol. 2007, 88 Pt 12, 3286–3293. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cladel, N.M.; Balogh, K.; Budgeon, L.; Christensen, N.D. Impact of genetic changes to the CRPV genome and their application to the study of pathogenesis in vivo. Virology 2007, 358, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, C.; Wettstein, F.O. The late region differentially regulates the in vitro transformation by cottontail rabbit papillomavirus DNA in different cell types. Virology 1991, 181, 637–646. [Google Scholar] [CrossRef]
- Brandsma, J.L. The cottontail rabbit papillomavirus model of high-risk HPV-induced disease. Methods Mol. Med. 2005, 119, 217–235. [Google Scholar] [PubMed]
- Holmgren, S.C.; Patterson, N.A.; Ozbun, M.A.; Lambert, P.F. The minor capsid protein L2 contributes to two steps in the human papillomavirus type 31 life cycle. J. Virol. 2005, 79, 3938–3948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorbar, J. Model systems of human papillomavirus-associated disease. J. Pathol. 2016, 238, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Ingle, A.; Ghim, S.; Joh, J.; Chepkoech, I.; Bennett Jenson, A.; Sundberg, J.P. Novel laboratory mouse papillomavirus (MusPV) infection. Vet. Pathol. 2011, 48, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cladel, N.M.; Budgeon, L.R.; Balogh, K.K.; Christensen, N.D. The Mouse Papillomavirus Infection Model. Viruses 2017, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.Y.; Majerciak, V.; Uberoi, A.; Kim, B.H.; Gotte, D.; Chen, X.; Cam, M.; Lambert, P.F.; Zheng, Z.M. The full transcription map of mouse papillomavirus type 1 (MmuPV1) in mouse wart tissues. PLoS Pathog. 2017, 13, e1006715. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Uberoi, A.; McGregor, S.M.; Wei, T.; Ward-Shaw, E.; Lambert, P.F. A Novel In Vivo Infection Model To Study Papillomavirus-Mediated Disease of the Female Reproductive Tract. MBio 2019, 10, e00180-19. [Google Scholar] [CrossRef] [Green Version]
- Cladel, N.M.; Budgeon, L.R.; Cooper, T.K.; Balogh, K.K.; Christensen, N.D.; Myers, R.; Majerciak, V.; Gotte, D.; Zheng, Z.M.; Hu, J. Mouse papillomavirus infections spread to cutaneous sites with progression to malignancy. J. Gen. Virol. 2017, 98, 2520. [Google Scholar] [CrossRef]
- Cladel, N.M.; Budgeon, L.R.; Balogh, K.K.; Cooper, T.K.; Brendle, S.A.; Christensen, N.D.; Schell, T.D.; Hu, J. Mouse papillomavirus infection persists in mucosal tissues of an immunocompetent mouse strain and progresses to cancer. Sci. Rep. 2017, 7, 16932. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, M.E.; Lambert, P.F. Sexual transmission of murine papillomavirus (MmuPV1) in Mus musculus. Elife 2019, 8, e50056. [Google Scholar] [CrossRef]
- Uberoi, A.; Lambert, P.F. Rodent Papillomaviruses. Viruses 2017, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Joh, J.; Jenson, A.B.; Ingle, A.; Sundberg, J.P.; Ghim, S.J. Searching for the initiating site of the major capsid protein to generate virus-like particles for a novel laboratory mouse papillomavirus. Exp. Mol. Pathol. 2014, 96, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Cladel, N.M.; Balogh, K.K.; Budgeon, L.R.; Mejia, A.F.; Christensen, N.D. Papillomavirus particles assembled in 293TT cells are infectious in vivo. J. Virol. 2006, 80, 11381–11384. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Budgeon, L.R.; Cladel, N.M.; Balogh, K.; Myers, R.; Cooper, T.K.; Christensen, N.D. Tracking vaginal, anal and oral infection in a mouse papillomavirus infection model. J. Gen. Virol. 2015, 96, 3554–3565. [Google Scholar] [CrossRef]
- Joh, J.; Jenson, A.B.; Proctor, M.; Ingle, A.; Silva, K.A.; Potter, C.S.; Sundberg, J.P.; Ghim, S.J. Molecular diagnosis of a laboratory mouse papillomavirus (MusPV). Exp. Mol. Pathol. 2012, 93, 416–421. [Google Scholar] [CrossRef]
- Cladel, N.M.; Jiang, P.; Li, J.J.; Peng, X.; Cooper, T.K.; Majerciak, V.; Balogh, K.K.; Meyer, T.J.; Brendle, S.A.; Budgeon, L.R.; et al. Papillomavirus can be transmitted through the blood and produce infections in blood recipients: Evidence from two animal models. Emerg. Microbes Infect. 2019, 8, 1108–1121. [Google Scholar] [CrossRef] [Green Version]
- Pastrana, D.V.; Gambhira, R.; Buck, C.B.; Pang, Y.Y.; Thompson, C.D.; Culp, T.D.; Christensen, N.D.; Lowy, D.R.; Schiller, J.T.; Roden, R.B. Cross-neutralization of cutaneous and mucosal Papillomavirus types with anti-sera to the amino terminus of L2. Virology 2005, 337, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Cladel, N.M.; Budgeon, L.R.; Cooper, T.K.; Balogh, K.K.; Hu, J.; Christensen, N.D. Secondary infections, expanded tissue tropism, and evidence for malignant potential in immunocompromised mice infected with Mus musculus papillomavirus 1 DNA and virus. J. Virol. 2013, 87, 9391–9395. [Google Scholar] [CrossRef] [Green Version]
- Beziat, V.; Rapaport, F.; Hu, J.; Titeux, M.; Bonnet des Claustres, M.; Bourgey, M.; Griffin, H.; Bandet, E.; Ma, C.S.; Sherkat, R.; et al. Humans with inherited T cell CD28 deficiency are susceptible to skin papillomaviruses but are otherwise healthy. Cell 2021, 184, 3812–3828.e30. [Google Scholar] [CrossRef] [PubMed]
- Cladel, N.M.; Budgeon, L.R.; Balogh, K.K.; Cooper, T.K.; Hu, J.; Christensen, N.D. A novel pre-clinical murine model to study the life cycle and progression of cervical and anal papillomavirus infections. PLoS ONE 2015, 10, e0120128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, L.T.; Broker, T.R.; Steinberg, B.M. The natural history of human papillomavirus infections of the mucosal epithelia. Apmis 2010, 118, 422–449. [Google Scholar] [CrossRef]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorbar, J. Host control of human papillomavirus infection and disease. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 27–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillner, L.; Heino, P.; Moreno-Lopez, J.; Dillner, J. Antigenic and immunogenic epitopes shared by human papillomavirus type 16 and bovine, canine, and avian papillomaviruses. J. Virol. 1991, 65, 6862–6871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Gissmann, L.; Sun, X.Y.; Kanjanahaluethai, A.; Muller, M.; Doorbar, J.; Zhou, J. Sequence close to the N-terminus of L2 protein is displayed on the surface of bovine papillomavirus type 1 virions. Virology 1997, 227, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Day, P.M.; Yutzy, W.H.; Lin, K.Y.; Hung, C.F.; Roden, R.B. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J. Virol. 2003, 77, 3531–3541. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse | Week 4 | Week 8 | Week 12 | Week 19 | Week 23 |
|---|---|---|---|---|---|
| #1V | 2.9 × 106 | 3.3 × 106 | 1.6 × 106 | 2.98 × 106 | 3.9 × 106 |
| #2V | 2.2 × 106 | 1.5 × 106 | 1.8 × 106 | 3.48 × 106 | 4.3 × 106 |
| #3V | 2.9 × 106 | 1.1 × 106 | 9.9 × 105 | 2.97 × 106 | 4.2 × 106 |
| #1A | 2.7 × 106 | 6.5 × 105 | 9.1 × 105 | 7.4 × 105 | 2.6 × 106 |
| #2A | 2.6 × 106 | 1.3 × 106 | 1.1 × 106 | 1.49 × 106 | 2.5 × 106 |
| #3A | 2.5 × 106 | 1.3 × 106 | 1.2 × 106 | 6.36 × 105 | 2.7 × 106 |
| Mouse | Week 4 | Week 7 | Week 9 | Week 11 |
|---|---|---|---|---|
| #1V | 5.7 × 105 | 2.1 × 106 | 6.2 × 105 | 7.1 × 105 |
| #2V | 1.6 × 106 | 1.3 × 106 | 1.7 × 106 | 8.6 × 105 |
| #3V | 7.2 × 105 | 3.4 × 105 | 1.6 × 105 | 6.7 × 105 |
| #1A | 4.6 × 105 | ND | ND | ND |
| #2A | 9.0 × 105 | 6.1 × 105 | 7 × 102 | 7.6 × 104 |
| #3A | 5.2 × 105 | ND | 9.7 × 104 | ND |
| Mouse | Week 4 | Week 7 | Week 9 | Week 11 |
|---|---|---|---|---|
| #1V | 4 × 105 | 1.96 × 105 | 1.1 × 105 | 2.1 × 105 |
| #2V | 4.6 × 105 | 1.74 × 105 | 8.7 × 104 | 6.1 × 105 |
| #3V | 6.6 × 105 | ND | 6.7 × 104 | 1.1 × 105 |
| #1A | 3.7 × 105 | ND | 4.2 × 105 | 2.1 × 105 |
| #2A | 4 × 105 | 2.56 × 105 | ND | 3.9 × 105 |
| #3A | 7.3 × 105 | ND | ND | 1.6 × 105 |
| Mouse | Week 4 | Week 7 | Week 9 | W11 |
|---|---|---|---|---|
| #1V | 5 × 105 | ND | 7.5 × 104 | 3.5 × 105 |
| #2V | 5.2 × 105 | 8.76 × 105 | 8.3 × 105 | 1.5 × 106 |
| #3V | 6.6 × 105 | ND | 6.7 × 104 | 1.1 × 105 |
| #1A | 1 × 106 | ND | 8.5 × 104 | 2.4 × 105 |
| #2A | 4.0 × 105 | ND | ND | ND |
| #3A | 8.8 × 105 | ND | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brendle, S.; Li, J.J.; Cladel, N.M.; Shearer, D.A.; Budgeon, L.R.; Balogh, K.K.; Atkins, H.; Costa-Fujishima, M.; Lopez, P.; Christensen, N.D.; et al. Mouse Papillomavirus L1 and L2 Are Dispensable for Viral Infection and Persistence at Both Cutaneous and Mucosal Tissues. Viruses 2021, 13, 1824. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091824
Brendle S, Li JJ, Cladel NM, Shearer DA, Budgeon LR, Balogh KK, Atkins H, Costa-Fujishima M, Lopez P, Christensen ND, et al. Mouse Papillomavirus L1 and L2 Are Dispensable for Viral Infection and Persistence at Both Cutaneous and Mucosal Tissues. Viruses. 2021; 13(9):1824. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091824
Chicago/Turabian StyleBrendle, Sarah, Jingwei J. Li, Nancy M. Cladel, Debra A. Shearer, Lynn R. Budgeon, Karla K. Balogh, Hannah Atkins, Marina Costa-Fujishima, Paul Lopez, Neil D. Christensen, and et al. 2021. "Mouse Papillomavirus L1 and L2 Are Dispensable for Viral Infection and Persistence at Both Cutaneous and Mucosal Tissues" Viruses 13, no. 9: 1824. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091824