
Genetic Diversity and Molecular Epidemiology of Circulating Respiratory Syncytial Virus in Central Taiwan, 2008–2017


Abstract
:1. Introduction
2. Materials and Methods
2.1. RSV Isolates
2.2. RNA Extraction, cDNA Synthesis, and G Protein Sequencing
2.3. Phylogenetic and Phylodynamic Analysis
2.4. Protein Substitution Analysis, Selection Pressure, and Glycosylation Prediction
3. Results
3.1. Frequency of RSV Isolates and Genotype Distribution in Taiwan, 2008–2017
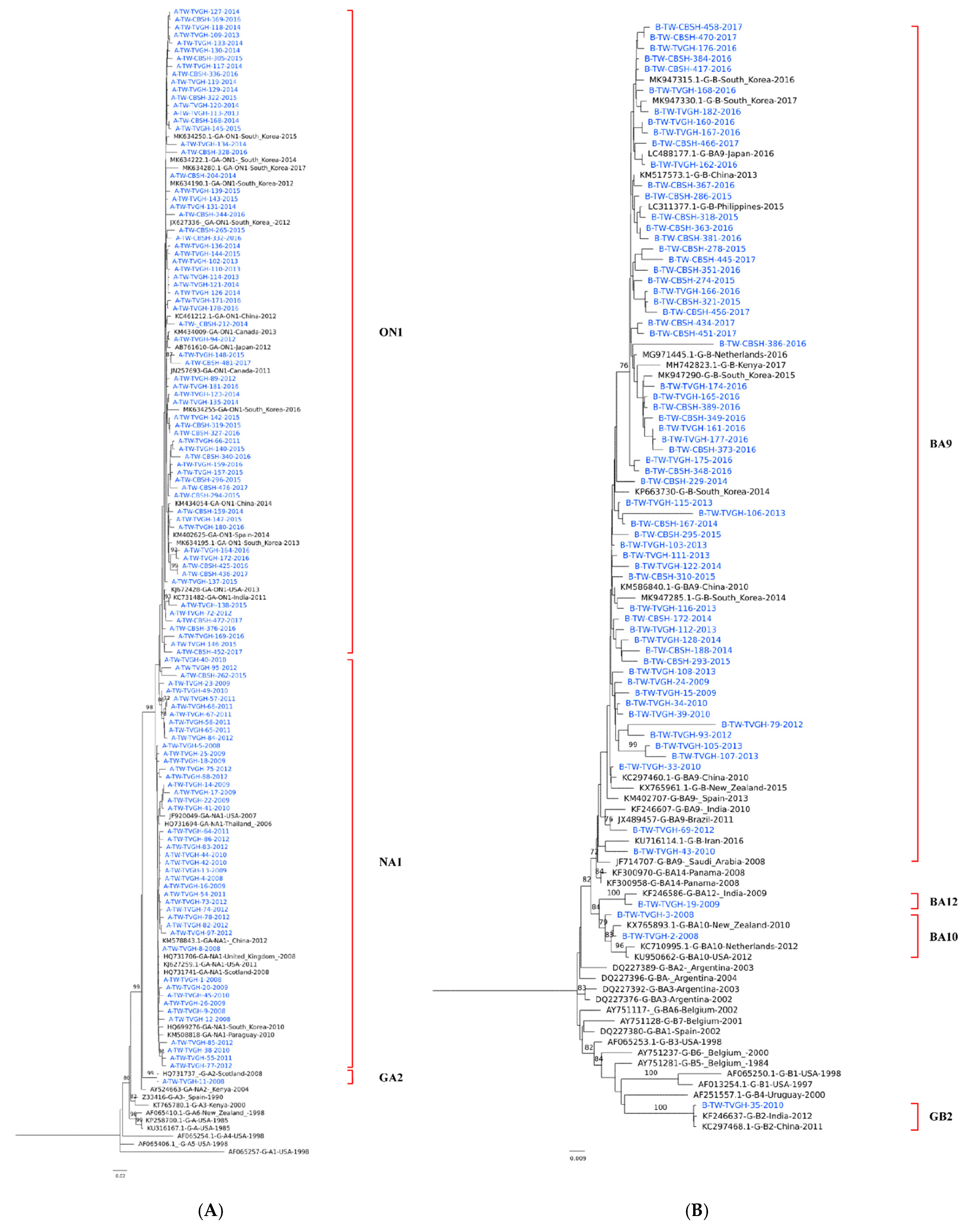
3.2. Phylogenetic Analysis of RSV
3.3. Deduced Amino Acid Analysis
3.4. Bayesian Skyline Plot and Evolution Rate
3.5. Selection Pressure Analysis
3.6. N-Linked and O-Linked Glycosylation Site Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jackson, M.L.; Scott, E.; Kuypers, J.; Nalla, A.K.; Roychoudury, P.; Chu, H.Y. Epidemiology of Respiratory Syncytial Virus Across Five Influenza Seasons among Adults and Children One Year of Age and Older-Washington State, 2011/2012–2015/2016. J. Infect. Dis. 2021, 223, 147–156. [Google Scholar] [CrossRef]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Akmar, L.Z.; Bailey, F.; Rath, B.A.; Alchikh, M.; Schweiger, B.; Lucero, M.G.; Nillos, L.T.; Kyaw, M.H.; Kieffer, A.; et al. Cost of Respiratory Syncytial Virus-Associated Acute Lower Respiratory Infection Management in Young Children at the Regional and Global Level: A Systematic Review and Meta-Analysis. J. Infect. Dis. 2020, 222 (Suppl. S7), S680–S687. [Google Scholar] [CrossRef]
- Do, L.A.H.; Wilm, A.; Van Doorn, H.R.; Lam, H.M.; Sim, S.; Sukumaran, R.; Tran, A.T.; Nguyen, B.H.; Tran, T.T.L.; Tran, Q.H.; et al. Direct whole-genome deep-sequencing of human respiratory syncytial virus A and B from Vietnamese children identifies distinct patterns of inter- and intra-host evolution. J. Gen. Virol. 2015, 96, 3470–3483. [Google Scholar] [CrossRef]
- Melero, J.A.; García-Barreno, B.; Martínez, I.; Pringle, C.R.; Cane, P.A. Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J. Gen. Virol. 1997, 78, 2411–2418. [Google Scholar] [CrossRef] [Green Version]
- Auksornkitti, V.; Kamprasert, N.; Thongkomplew, S.; Suwannakarn, K.; Theamboonlers, A.; Samransamruajkij, R.; Poovorawan, Y. Molecular characterization of human respiratory syncytial virus, 2010-2011: Identification of genotype ON1 and a new subgroup B genotype in Thailand. Arch. Virol. 2014, 159, 499–507. [Google Scholar] [CrossRef]
- Esposito, S.; Piralla, A.; Zampiero, A.; Bianchini, S.; Di Pietro, G.; Scala, A.; Pinzani, R.; Fossali, E.; Baldanti, F.; Principi, N. Characteristics and Their Clinical Relevance of Respiratory Syncytial Virus Types and Genotypes Circulating in Northern Italy in Five Consecutive Winter Seasons. PLoS ONE 2015, 10, e0129369. [Google Scholar]
- Hibino, A.; Saito, R.; Taniguchi, K.; Zaraket, H.; Shobugawa, Y.; Matsui, T.; Suzuki, H.; for the Japanese HRSV Collaborative Study Group. Molecular epidemiology of human respiratory syncytial virus among children in Japan during three seasons and hospitalization risk of genotype ON1. PLoS ONE 2018, 13, e0192085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malasao, R.; Okamoto, M.; Chaimongkol, N.; Imamura, T.; Tohma, K.; Dapat, I.; Dapat, C.; Suzuki, A.; Saito, M.; Saito, M.; et al. Molecular Characterization of Human Respiratory Syncytial Virus in the Philippines, 2012–2013. PLoS ONE 2015, 10, e0142192. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Park, P.H.; Huh, J.W.; Yun, H.J.; Lee, H.K.; Yoon, M.H.; Lee, S.; Ko, G. Molecular and clinical characterization of human respiratory syncytial virus in South Korea between 2009 and 2014. Epidemiol. Infect. 2017, 145, 3226–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretorius, M.; Van Niekerk, S.; Tempia, S.; Moyes, J.; Cohen, C.; Madhi, S.A.; Venter, M. Replacement and Positive Evolution of Subtype A and B Respiratory Syncytial Virus G-Protein Genotypes From 1997–2012 in South Africa. J. Infect. Dis. 2013, 208 (Suppl. S3), S227–S237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshaghi, A.; Duvvuri, V.; Lai, R.; Nadarajah, J.T.; Li, A.; Patel, S.N.; Low, D.E.; Gubbay, J.B. Genetic Variability of Human Respiratory Syncytial Virus A Strains Circulating in Ontario: A Novel Genotype with a 72 Nucleotide G Gene Duplication. PLoS ONE 2012, 7, e32807. [Google Scholar] [CrossRef] [Green Version]
- Trento, A.; Galiano, M.; Videla, C.; Carballal, G.; García-Barreno, B.; Melero, J.A.; Palomo, C. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J. Gen. Virol. 2003, 84, 3115–3120. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Hsiao, K.-L.; Weng, L.-C.; Liu, C.-P.; Liu, H.-F. Persistence and continuous evolution of the human respiratory syncytial virus in northern Taiwan for two decades. Sci. Rep. 2019, 9, 4704. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, A.C.; Lebbink, R.J.; Naaktgeboren, C.; Evers, A.; Viveen, M.C.; Greenough, A.; Heikkinen, T.; Stein, R.T.; Richmond, P.; Martinón-Torres, F.; et al. Global molecular diversity of RSV—The “INFORM RSV“ study. BMC Infect. Dis. 2020, 20, 450. [Google Scholar] [CrossRef]
- Datamonkey Adaptive Evolution Server. Available online: https://www.datamonkey.org/GARD (accessed on 22 April 2021).
- Yun, K.W.; Choi, E.H.; Lee, H.J. Molecular epidemiology of respiratory syncytial virus for 28 consecutive seasons (1990–2018) and genetic variability of the duplication region in the G gene of genotypes ON1 and BA in South Korea. Arch. Virol. 2020, 165, 1069–1077. [Google Scholar] [CrossRef]
- Dapat, I.C.; Shobugawa, Y.; Sano, Y.; Saito, R.; Sasaki, A.; Suzuki, Y.; Kumaki, A.; Zaraket, H.; Dapat, C.; Oguma, T.; et al. New Genotypes within Respiratory Syncytial Virus Group B Genotype BA in Niigata, Japan. J. Clin. Microbiol. 2010, 48, 3423–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thongpan, I.; Mauleekoonphairoj, J.; Vichiwattana, P.; Korkong, S.; Wasitthankasem, R.; Vongpunsawad, S.; Poovorawan, Y. Respiratory syncytial virus genotypes NA1, ON1, and BA9 are prevalent in Thailand, 2012–2015. PeerJ Comput. Sci. 2017, 5, e3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahombanjanahary, N.H.R.; Rybkina, K.; Randriambolamanantsoa, T.H.; Razafimanjato, H.; Heraud, J.M. Genetic diversity and molecular epidemiology of respiratory syncytial virus circulated in Antananarivo, Madagascar, from 2011 to 2017: Predominance of ON1 and BA9 genotypes. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2020, 129, 104506. [Google Scholar]
- Duvvuri, V.R.; Granados, A.; Rosenfeld, P.; Bahl, J.; Eshaghi, A.; Gubbay, J.B. Genetic diversity and evolutionary insights of respiratory syncytial virus A ON1 genotype: Global and local transmission dynamics. Sci. Rep. 2015, 5, srep14268. [Google Scholar]
- Yoshihara, K.; Le, M.N.; Nagasawa, K.; Tsukagoshi, H.; Nguyen, H.A.; Toizumi, M.; Moriuchi, H.; Hashizume, M.; Ariyoshi, K.; Dang, D.A.; et al. Molecular evolution of respiratory syncytial virus subgroup A genotype NA1 and ON1 attachment glycoprotein (G) gene in central Vietnam. Infect. Genet. Evol. 2016, 45, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Xia, Q.; Xiao, Q.; Zhou, L.; Zang, N.; Long, X.; Xie, X.; Deng, Y.; Wang, L.; Fu, Z.; et al. The genetic variability of glycoproteins among respiratory syncytial virus subtype A in China between 2009 and 2013. Infect. Genet. Evol. 2014, 27, 339–347. [Google Scholar] [CrossRef]
- Otieno, J.R.; Kamau, E.M.; Agoti, C.N.; Lewa, C.; Otieno, G.; Bett, A.; Ngama, M.; Cane, P.A.; Nokes, D.J. Spread and Evolution of Respiratory Syncytial Virus A Genotype ON1, Coastal Kenya, 2010–2015. Emerg. Infect. Dis. 2017, 23, 264–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamau, E.; Otieno, J.R.; Lewa, C.S.; Mwema, A.; Murunga, N.; Nokes, D.J.; Agoti, C.N. Evolution of respiratory syncytial virus genotype BA in Kilifi, Kenya, 15 years on. Sci. Rep. 2020, 10, 21176. [Google Scholar] [CrossRef]
- Di Giallonardo, F.; Kok, J.; Fernandez, M.; Carter, I.; Geoghegan, J.L.; Dwyer, D.E.; Holmes, E.C.; Eden, J.-S. Evolution of Human Respiratory Syncytial Virus (RSV) over Multiple Seasons in New South Wales, Australia. Viruses 2018, 10, 476. [Google Scholar] [CrossRef] [Green Version]
- Haider, S.H.; Khan, W.H.; Deeba, F.; Ali, S.; Ahmed, A.; Naqvi, I.H.; Dohare, R.; Alsenaidy, H.A.; Alsenaidy, A.M.; Broor, S.; et al. BA9 lineage of respiratory syncytial virus from across the globe and its evolutionary dynamics. PLoS ONE 2018, 13, e0193525. [Google Scholar]
- Fall, A.; Elawar, F.; Hodcroft, E.B.; Jallow, M.M.; Toure, C.T.; Barry, M.A.; Kiori, D.E.; Sy, S.; Diaw, Y.; Goudiaby, D.; et al. Genetic diversity and evolutionary dynamics of respiratory syncytial virus over eleven consecutive years of surveillance in Senegal. Infect. Genet. Evol. 2021, 91, 104864. [Google Scholar] [CrossRef] [PubMed]
- Schobel, S.; Stucker, K.; Moore, M.L.; Anderson, L.J.; Larkin, E.K.; Shankar, J.; Bera, J.; Puri, V.; Shilts, M.H.; Rosas-Salazar, C.; et al. Respiratory Syncytial Virus whole-genome sequencing identifies convergent evolution of sequence duplication in the C-terminus of the G gene. Sci. Rep. 2016, 6, 26311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabatabai, J.; Prifert, C.; Pfeil, J.; Grulich-Henn, J.; Schnitzler, P. Novel respiratory syncytial virus (RSV) genotype ON1 predominates in Germany during winter season 2012–2013. PLoS ONE 2014, 9, e109191. [Google Scholar] [CrossRef] [Green Version]
- Harvey, W.T.; Carabelli, A.; Jackson, B.; Gupta, R.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 |
|---|---|---|---|---|---|---|---|---|---|---|
| Total | 10 | 13 | 15 | 11 | 21 | 15 | 29 | 32 | 52 | 15 |
| RSV A | ||||||||||
| GA2 | 1 (10) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| NA1 | 6 (60) | 10 (77) | 9 (60) | 10 (91) | 15 (71) | 0 | 0 | 2 (6) | 0 | 0 |
| ON1 | 0 | 0 | 0 | 1 (9) | 3 (14) | 6 (40) | 22 (76) | 21 (66) | 20 (38) | 6 (40) |
| RSV B | ||||||||||
| GB2 | 0 | 0 | 1 (7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BA9 | 1 (10) | 2 (15) | 4 (27) | 0 | 3 (14) | 9 (60) | 7 (24) | 9 (28) | 32 (62) | 9 (60) |
| BA10 | 2 (20) | 0 | 1 (7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BA12 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Genotype | SLAC | FEL | FUBAR | |||
|---|---|---|---|---|---|---|
| dN/dS Mean | Amino Acid Substitution * | dN/dS Mean | Amino Acid Substitution | BayesFactor | Amino Acid Substitution | |
| NA1 | 0.657 | L274P | 0.647 | L274P ** | 25.152 | L274P |
| ON1 | 0.625 | L274P | 0.616 | L274P | 27.933 | L274P |
| L298P | L298P | 57.545 | L298P | |||
| BA9 | 0.468 | H287Y ** | 0.455 | H287Y | 79.643 | H287Y |
| T270I ** T270F ** | T312N T312A T312I | 97.297 | T312N T312A T312I | |||
| Genotype | Putative N-Glycosylation Site % (n/N) | ||
|---|---|---|---|
| NA1 | N266 | 2.2% | (1/45) |
| N237 | 4.4% | (2/45) | |
| N251 | 91.1% | (41/45) | |
| N294 | 60% | (27/45) | |
| ON1 | N237 | 100.0% | (68/68) |
| N318 | 95.6% | (65/68) | |
| N242 | 1.5% | (1/68) | |
| BA9 | N296 | 98.3% | (59/60) |
| N310 | 86.7% | (52/60) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-Y.; Fang, Y.-P.; Wang, L.-C.; Chou, T.-Y.; Liu, H.-F. Genetic Diversity and Molecular Epidemiology of Circulating Respiratory Syncytial Virus in Central Taiwan, 2008–2017. Viruses 2022, 14, 32. https://0-doi-org.brum.beds.ac.uk/10.3390/v14010032
Lee C-Y, Fang Y-P, Wang L-C, Chou T-Y, Liu H-F. Genetic Diversity and Molecular Epidemiology of Circulating Respiratory Syncytial Virus in Central Taiwan, 2008–2017. Viruses. 2022; 14(1):32. https://0-doi-org.brum.beds.ac.uk/10.3390/v14010032
Chicago/Turabian StyleLee, Chun-Yi, Yu-Ping Fang, Li-Chung Wang, Teh-Ying Chou, and Hsin-Fu Liu. 2022. "Genetic Diversity and Molecular Epidemiology of Circulating Respiratory Syncytial Virus in Central Taiwan, 2008–2017" Viruses 14, no. 1: 32. https://0-doi-org.brum.beds.ac.uk/10.3390/v14010032