1. Introduction
Infectious bovine rhinotracheitis (IBR), also known as necrotizing rhinitis and red nose disease, is a respiratory contact infection of cattle caused by bovine herpesvirus type 1 (BHV−1). Its clinical manifestations are varied, including respiratory tract changes, conjunctivitis, abortion, mastitis, and induced calf encephalitis. This disease is currently prevalent in the world and has a significant impact on milk yield of dairy cattle, fecundity of bulls, and servitude of draft cattle [
1,
2]. It belongs to the family Herpesviridae and subfamily Alphaherpesvirinae. The members of this family are large, enveloped, and double−stranded DNA viruses, and among them, BHV−1 is an important member that causes severe economic losses to the cattle industry worldwide [
3,
4].
Long noncoding RNA (lncRNA) is a class of noncoding RNA with a length of more than 200 nt, transcribed by RNA polymerase II or III, existing in the nucleus and cytoplasm, with long transcripts and lack of open reading frame, and unable to translate proteins in cells [
5]. They participate in gene regulation in the form of RNA in epigenetic regulation, transcriptional regulation, and posttranscriptional regulation [
6]. Studies have shown that lncRNA is closely related to tissue differentiation, organogenesis, and tumorigenesis [
7]. In addition, lncRNA is also closely related to virus infection, and it has been found that many viruses, such as human immunodeficiency virus (HIV), human hepatitis B virus (HBV), human hepatitis C virus (HCV), and avian bursa of bursa virus (IBDV) can induce cells to produce lncRNAs. This lncRNA production plays a key role in viral replication, interferon production, NF−κB, STAT, and other pathways [
8,
9,
10,
11], but it has not been reported whether similar lncRNA is produced after BHV−1 infection.
To understand the mechanism of lncRNA in BHV−1 infection, bovine kidney subculture (MDBK) cells were infected with BHV−1 in this study. Transcriptome sequencing in separate phases was conducted to obtain the lncRNA expression profile of BHV−1−infected MDBK cells [
12], a novel and upregulated lncRNA−
MSTRG.16919.1 was screened out, but the function of lncRNA−
MSTRG.16919.1 is unknown, so transcriptome sequencing was performed to understand the function of the lncRNA−
MSTRG.16919.1 gene after it was silenced.
2. Materials and Methods
2.1. Experimental Materials
Bovine herpesvirus type 1 (BHV−1) and MDBK were stored in the cell biology laboratory of Heilongjiang Bayi Agricultural University (Daqing, China). The Ribo FECTTMCP transfection reagent and Ribo lncRNA smart silencer interfering plasmid were designed and synthesized by Guangzhou RiboBio. RNA extraction, cDNA reverse transcription, and fluorescence quantitative PCR kits were all purchased from Nanjing Vazyme Biotechnology. A BCA protein concentration determination kit and RIPA lysate were purchased from Solarbio. Beta actin and sheep anti−mouse IgG−HRP antibodies were purchased from Bioss. NF−κB, JNK and IκB antibodies were purchased from Affinity Biosciences.
2.2. Sample Preparation
The MDBK cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (Biological Industries, Kibbutz Beit−Haemek, Israel) and cultured in a 5% CO2 incubator at 37 °C. On the day before transfection, 1 × 106 cells were inoculated in a 6−well plate culture well containing a proper amount of complete medium consisting of 10% FBS with 1% penicillin and streptomycin dual antibiotic, so that the cell density during transfection reached 80%. The experimental groups were as follows. The siRNA−MSTRG.16919.1 group was the experimental group, in which MDBK cells were infected with BHV−1 for 33 h and silenced lncRNA−MSTRG.16919.1; siRNA−MSTRG.16919.1 NC group was MDBK cells infected with BHV−1 for 33 h and silenced an unrelated sequence was the negative control group of the siRNA−MSTRG.16919.1 group; the BHV−1 33 h group was the MDBK cells infected with BHV−1 33 h as the reference group; and the blank group was the MDBK cell group without any treatment. BHV−1 was inoculated into monolayer MDBK cells at MOI (multiplicity of infection) = 1 dose and then transfected after 1 h.
RiboFECT™ CP buffer (10×) was diluted with sterile phosphate buffer saline (PBS) to prepare RiboFECT CP Buffer (1×). RiboFECT CP reagent was removed from 4 °C storage and fully oscillated in a vortex oscillator at room temperature. A 20 μL 20 μM smart silencer siRNA storage solution was diluted with 120 μL 1× riboFECT CP buffer, mixed gently, and incubated at room temperature for 5 min. Next, 12 μL riboFECT CP reagent was added, gently blown, and mixed and incubated at room temperature for up to 15 min. After incubation, the RiboFECT CP mixture was added to 1848 μL cell culture medium, gently mixed, and the culture plate cultured in a carbon dioxide incubator at 37 °C. After 18 h of infection, the occurrence of cytopathy (CPE) was observed and photos taken every hour. When cracks were observed in the cell layer, the samples were collected and washed with Hank’s solution, then digested with Trizol. Finally, the samples were collected into a nuclease−free cell cryopreservation tube, sealed with a sealing membrane, and frozen in liquid nitrogen. One part of the samples was used to verify the gene silencing effect. After lncRNA−MSTRG.16919.1 silencing was confirmed, the other part of the samples was transported on dry ice to Guangzhou Gidio Biotechnology for high−throughput sequencing.
2.3. RNA−seq
Total RNA was extracted from siRNA−MSTRG.16919.1 samples, siRNA−MSTRG.16919.1NC negative control samples, and BHV−33 h samples. The RNA quality was evaluated on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and examined using agarose gel electrophoresis without RNase. After extracting total RNA, eukaryotic mRNA was enriched with oligomer (DT) beads. The enriched mRNA was cut into short fragments with fragment buffer and reverse−transcribed into cDNA with NEBNext Ultra RNA Library Prep Kit for Illumina (NEB#7530, New England Biolabs, Ipswich, MA, USA). The purified double−stranded cDNA fragments were end repaired, A base added, and ligated to Illumina sequencing adapters. The ligation reaction was purified with the AMPure XP Beads (1.0X). Ligated fragments were subjected to size selection by agarose gel electrophoresis and polymerase chain reaction (PCR) amplified. The resulting cDNA library was sequenced using Illumina Novaseq6000 by Gene Denovo Biotechnology (Guangzhou, China). Reads obtained from the sequencing machines included raw reads containing adapters or low−quality bases that would affect the following assembly and analysis. Thus, to get high−quality clean reads, reads were further filtered by fastp (version 0.18.0). The steps for read filtering involved removing reads with adapters, reads containing more than 10% N, reads that were all A bases and of low quality, where the base number of mass value Q ≤ 20 accounted for more than 50% of the whole read.
2.4. Statistics on the Number of Differentially Expressed mRNA
The bioinformatic software SIMCA 14.1 (Umetrics, Sverige) was used to analyze the sequencing data and differentially expressed mRNA. The screening criteria of differentially expressed mRNA were |log
2(FC)| > 1 and FDR < 0.05. Principal component analysis (PCA) was used to analyze and calculate the Pearson’s correlation coefficient between samples, to understand the repeatability between samples and assist in excluding outliers. According to significantly different mRNA in each comparison group, volcanic map analysis was conducted to visually show the different mRNA among the comparison groups. mRNA with similar expression patterns may have common functions or participate in common metabolic pathways and signal pathways, so the analysis was mainly divided into three parts [
13]: normalize the read count, calculate the probability of hypothesis test (
p value) according to the model, and carry out multiple hypothesis tests and corrections to obtain the error−detection rate (FDR). The read count was normalized, the probability of hypothesis test (
p value) calculated according to the model. and then multiple hypothesis tests and corrections conducted to obtain FDR values or error−detection rates.
2.5. Differentially Expressed mRNA by Real−Time Quantitative PCR (RT−qPCR)
According to the sequencing results, three differential genes were randomly selected for upregulation and downregulation. The primers used in RT−qPCR were designed by Primer Premier 5 (Premier, British Columbia, BC, Canada) software and compared by NCBI (National Institutes of Health, Bethesda, MD, USA). All primers were synthesized by Shanghai Sangon Bioengineering Technology Service (
Table 1). Trizol (Ambion, Austin, TX, USA) reagent was used to extract total RNA from cells and the concentration and quality of extracted RNA were determined by NanoDrop 2000 (Thermo, Waltham, MA, USA) A reverse−transcription kit (Vazyme, Jiangsu, China) was used to synthesize cDNA strands, and cDNA was used as template to detect differentially expressed genes by RT−qPCR to verify the accuracy of sequencing. Ubiquitin C−terminal hydrolase L5 [
14] was selected as the internal reference gene and the
t−test and 2−ΔΔCt method were used to analyze the data of the relative transcription level of each gene. Three multiple holes were set for each gene in each experiment and three independent repeated experiments were conducted. Finally, the average of relative transcription level was calculated. The relative transcription level of each gene was calculated by the following formula: mRNA relative transcription expression = 2−ΔΔCT, ΔCt value = target gene Ct value−internal reference gene Ct value. The results of RT−qPCR were analyzed by GraphPad. Prism, with *
p < 0.05, **
p < 0.01, ***
p < 0.001, ****
p < 0.0001.
2.6. GO Enrichment Analysis of Target Gene
The differential genes were mapped to each term of the GO database (GO database. Available online:
http://www.geneontology.org/ (accessed on 12 June 2022)) [
15] and the number of differential genes in each term calculated to get the statistics of the number of differential genes in the list of differential genes with a certain GO function. The hypergeometric test was then used to find out the GO items that were significantly enriched in the differential genes compared with the background [
16]. After the calculated
p value was corrected by Bonferroni, the threshold value was corrected with
p values less than 0.05 and the GO term satisfying this condition was defined as a GO term that was significantly enriched in differentially expressed proteins. The main biological functions of differentially expressed proteins were determined by GO functional significance enrichment analysis [
17].
2.7. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of Target Genes
Pathway significant enrichment analysis is based on KEGG Pathway as a unit and a hypergeometric test is used to find the pathways that are significantly enriched in differential genes compared with the whole background. Through pathway significance enrichment, the most important biochemical metabolic pathways and signal transduction pathways involved in differential genes can be determined. The calculated p value was corrected by FDR and FDR less than 0.05 was taken as the threshold. Pathways satisfying this condition were defined as significantly abundant pathways in differentially expressed genes (DEGs).
2.8. Western Blot Verification of Partial Signal Pathways of KEGG Enrichment
According to the signal pathways obtained from KEGG enrichment analysis, some signal pathways related to virus infection were verified by Western blots. The sample was lysed with high−efficiency RIPA cell lysate (Solarbio, Beijing, China). The sample was placed on ice for lysis for 30 min and tapped gently every 10 min. The cells were treated with a cell scraper and transferred to a centrifuge tube and centrifuged for 5 min at 4 °C at 12,000 rpm/min and the supernatant was subpackaged and stored. The protein was quantified by a BCA protein quantitative kit (Solarbio, Beijing, China) and its concentration determined.
PAGE separation gel and concentrated gel were prepared according to the polyacrylamide gel formula (Solarbio, Beijing, China). After electrophoresis at 80 V for 30 min, the voltage was switched to 120 V and then electrophoresis conducted for 1 h. After electrophoresis, the concentrated gel on the gel was removed. The filter paper, nitrocellulose membrane (NC), gel and filter paper were soaked in pre−membrane transfer solution and sequentially placed in a wet transfer tank and bubbles in the filter paper rolled out by a glass rod. After installation, the transfer slot was covered, the power supply turned on at 200 mA constant current and the film rotated for 2 h in an ice bath. After tris buffered saline tween (TBST) cleaning, the transformed NC membrane was soaked in 5% skimmed milk and sealed at room temperature for 2 h. Skimmed milk was removed, and the NC membrane was washed with TBST three times, then 1:1000 diluted primary antibody was added and incubated overnight at 4 °C. The NC membrane was cleaned again with TBST three times, 1:5000 diluted goat anti−rabbit IgG−HRP antibody was added and incubated at room temperature for 1 h. The NC film was then cleaned with TBST three times, then ECL (Merck KGaA, Darmstadt, Germany) chromogenic solution was used to take pictures with an AI600 imaging system and ImageJ image analysis software was used to quantitatively analyze protein.
2.9. Protein Interaction Network Analysis
The interaction relationships in the STRING [
18] protein interaction database (STRING. Available online:
http://string−db.org/ (accessed on 12 June 2022)) [
19] were used to analyze the differential gene−protein interaction network. Aiming at the species contained in the database, the differential gene set was extracted from the database and Cytoscape [
20] was used to construct the interaction relationship network diagram. For the species not included in the database, the sequence of the target gene set was first aligned with the protein sequence of the reference species contained in the string database by blastx and the interaction network constructed by using the protein interaction relationship of the reference species in the alignment.
4. Discussion
Bovine herpes virus type I (BHV−1) is a member of the alpha herpesvirus subfamily, which can cause many diseases in bovines by proliferating in MDBK cells and causing cytopathic changes. The replication and growth characteristics of BHV−1 have been understood, but the pathogenesis of BHV−1 and the interaction between virus and host needed to be further studied to assist the treatment and prevention of bovine infectious rhinotracheitis.
As a multifunctional regulator of transcription, lncRNA not only participates in the epigenetic control of transcription initiation to the regulation of mature transcription stability, but also participates in many life activities, such as innate immune response and host–virus interaction regulation. Virus infection can cause differential expression of lncRNA in cells and differentially expressed lncRNA can regulate innate immune response in many ways, resisting virus infection. Studies have shown that in A549 cells infected with influenza A virus (IAV), lncRNA−IVRIPE, which inhibits IAV by promoting interferon, and interferon−stimulating gene expression had a significantly increasing trend, while overexpression of IVRIPE inhibits IAV replication [
23] and IVRIPE is an important regulator of host antiviral response. Some studies have shown that viruses can negatively regulate the host’s antiviral immune response by using lncRNA encoded by themselves and their hosts, creating an environment for their own replication, so 3’−untranslated region (3’ UTR) of flavivirus genome can transcribe small ncRNA, or flavivirus subgenomic RNA (sfRNA). The lncRNA protects viral RNA from host exoribonuclease 1 (XRN1) degradation in infected cells [
24].
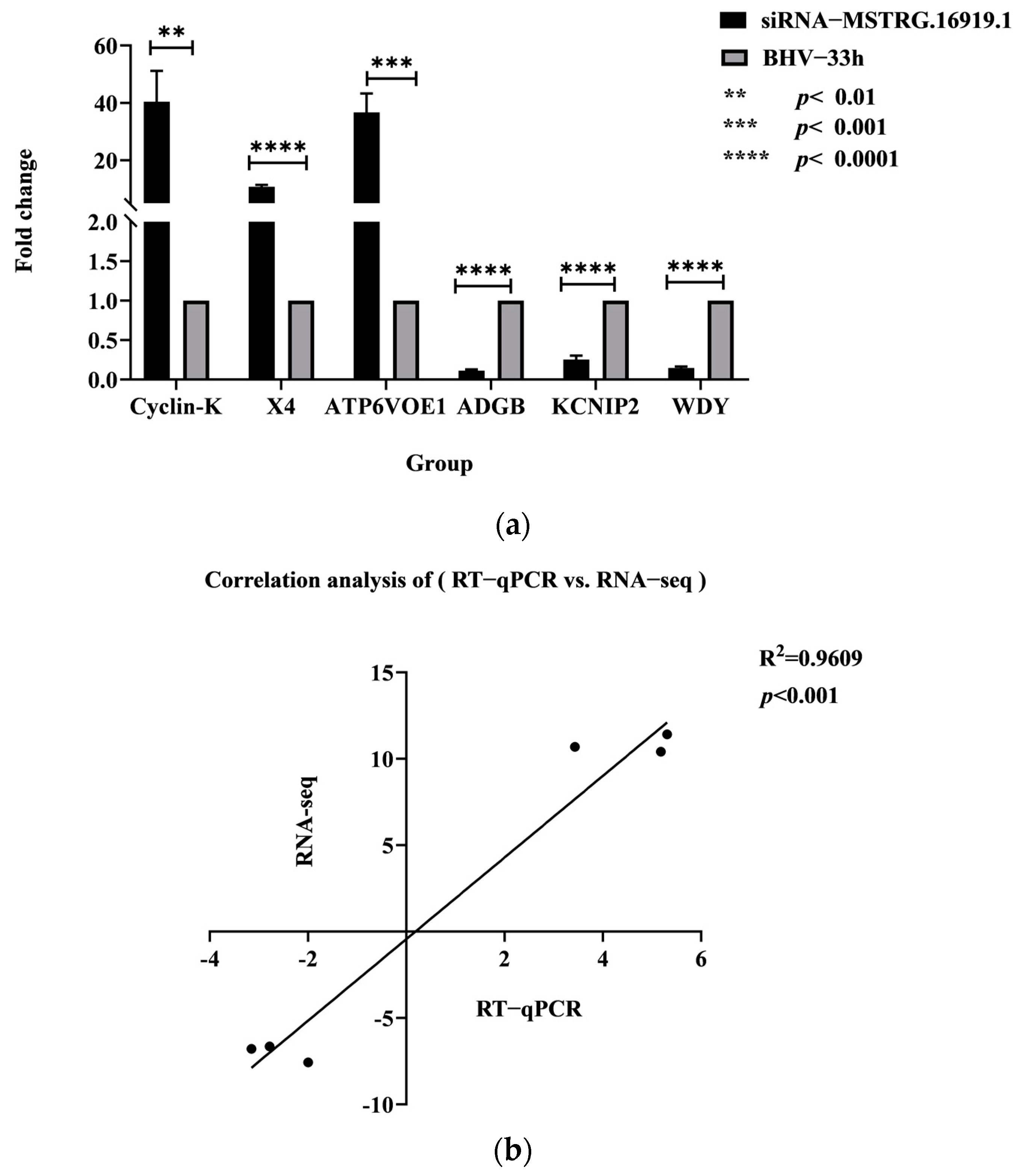
The lncRNA−MSTRG.16919.1 is a novel lncRNA that was significantly increased in BHV−1 infected MDBK cells by whole−transcription sequencing and analysis, but its function remained unclear. There is no research reported on the analysis of mRNA associated with lncRNA−MSTRG.16919.1 in MDBK cells infected by BHV−1, because its genome is too large to be overexpressed. The lncRNA−MSTRG.16919.1 gene was therefore silenced and transcriptome sequenced to explore the role of lncRNA−MSTRG.16919.1 in the process of BHV−1 infecting MDBK cells. Since it was uncertain whether lncRNA−MSTRG.16919.1 was in the nucleus or cytoplasm, the techniques of RNA interference (RNAi) and (antisense oligonucleotide ASO) were used to silence them as RNAi targets lncRNA in the cytoplasm while ASO targets lncRNA in the nucleus. The sequencing data were analyzed by EdgeR software, and the differential genes generated by the siRNA−MSTRG.16919.1 and siRNA−MSTRG.16919.1 NC group and the siRNA−MSTRG.16919.1 and BHV−1 33 h group were screened, and a volcano plot and 498 repetitive mRNA genes were obtained. Six differentially expressed mRNA were then selected from repeated genes including three upregulated and three downregulated genes, which were verified by RT−qPCR. The change trend of gene expression was consistent with the sequencing results, which indicated that the accuracy of this sequencing result could be used for subsequent experimental study of downstream functions.
To determine the biological processes of mRNA associated with lncRNA−MSTRG.16919.1, GO and KEGG enrichment analyses were conducted on mRNA associated with lncRNA−MSTRG.16919.1. The results of GO enrichment analysis showed that the biological processes of mRNA associated with lncRNA−MSTRG.16919.1 were molecular functions including nucleotide, purine nucleoside and enzyme binding, which indicated that lncRNA−MSTRG.16919.1 had the above functions. In cellular components, it involved most of the intracellular and cytoskeleton parts, organelles, microtubule cytoskeleton, microtubule tissue center and spindle microtubules, which indicated that this lncRNA participated in the synthesis of cell components. In biological processes, it included cell cycle and glial macromolecule metabolism processes, which showed that lncRNA−MSTRG.16919.1 was mostly involved in the above biological processes.
The results of KEGG enrichment analysis showed that lncRNA−MSTRG.16919.1 was involved in cancer, viral infection, cell cycle, apoptosis and other pathways. Among them, Toll and Imd signaling, TNF signaling, NF−κB signaling and MAPK signaling pathways were related to virus infection. The results of GO and KEGG enrichment analyses suggested that lncRNA−MSTRG.16919.1 was a multifunctional lncRNA and played an important regulatory role in a variety of biological processes. The NF−κB, IκB and JNK proteins were detected in the NF−κB signaling and MAPK signaling pathways by Western blot. The results showed that the expression of NF−κB protein and JNK protein was downregulated, while the expression of IκB protein was increased in the siRNA−MSTRG.16919.1 group compared with the siRNA−MSTRG.16919.1NC group, BHV−33 h group, and MDBK group. The above data indicated that lncRNA−MSTRG.16919.1 may participate in this signaling pathway and then participate in the regulation of virus infection.
Protein interaction networks encoded by differential genes were constructed using the STRING protein interaction database and the top 20 proteins including RANBP2, KDM6A, ATAD2, PIK3CA, STAG2, DMXL1, CE3P50, BTAF1, PTEN, SKIV2L2, LTN1, BBIRC6, USP34, CLTC, SCAF11, PIK3CB, MAPK8, TRIP12, PDS5A and JAK2 were further screened by Cytoscape. These results confirmed that lncRNA−
MSTRG.16919.1 was a multifunctional lncRNA, which participated in apoptosis by BIRC6, pyroptosis by SCAF11, tumor suppressor and metabolic regulator using PTEN, formation of nuclear pore complexes using RANBP2, viral infection and cellular immunity using SKIV2L2, JAK2, PIK3CB and MAPK8. It has been found that in SKIV2L−deficient cells, the unfolded protein response (UPR) produced an endogenous RLR ligand through the IRE−1 endonuclease lysis of cell RNA, which triggered the production of type I interferon (IFN) and that SKIV2L RNA exocrine can effectively limit the activation of RLRs. Studies have shown that JAK2−V617F promoted the synthesis of PD−L1 in MPN cells, which led to the limitation of the antitumor T cell response and the change of T cell metabolism and finally led to JAK2−V617F driving the immune escape of MPN cells [
25].
The JAK2 and IL6/JAK2/STAT3 signaling pathways are therapeutic targets for treating excessive inflammatory response to virus infection, where PIK3CB, the target of miRNA−34a participated in TCR−mediated NF−κB signaling after binding to B7 ligand on antigen presenting cells (APC). This molecular interaction activated the PIK3 complex, triggered phosphorylation of PRKCQ through pyruvate dehydrogenase kinase 1 and led to downstream activation of NF−κB [
26].
The MAPK8 gene is also known as JNK and has been identified as being affected by viral infection. Using an overexpression experiment, Fung showed that JNK/MAPK8 participated in coronavirus–host interaction [
27], so silencing lncRNA
−MSTRG.16919.1 may affect the expression of SKIV2L2, JAK2, PIK3CB and MAPK8 proteins and then regulate the infection of MDBK cells by BHV−1.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}