
Increased Heparanase Levels in Urine during Acute Puumala Orthohantavirus Infection Are Associated with Disease Severity

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Clinical Samples
2.2. HPSE Measurement Assay
2.3. ELISAs
2.4. Virus Isolates
2.5. Podocyte Cultures
2.6. Immunofluorescence
2.7. HPSE mRNA Expression
2.8. Statistical Analysis
3. Results
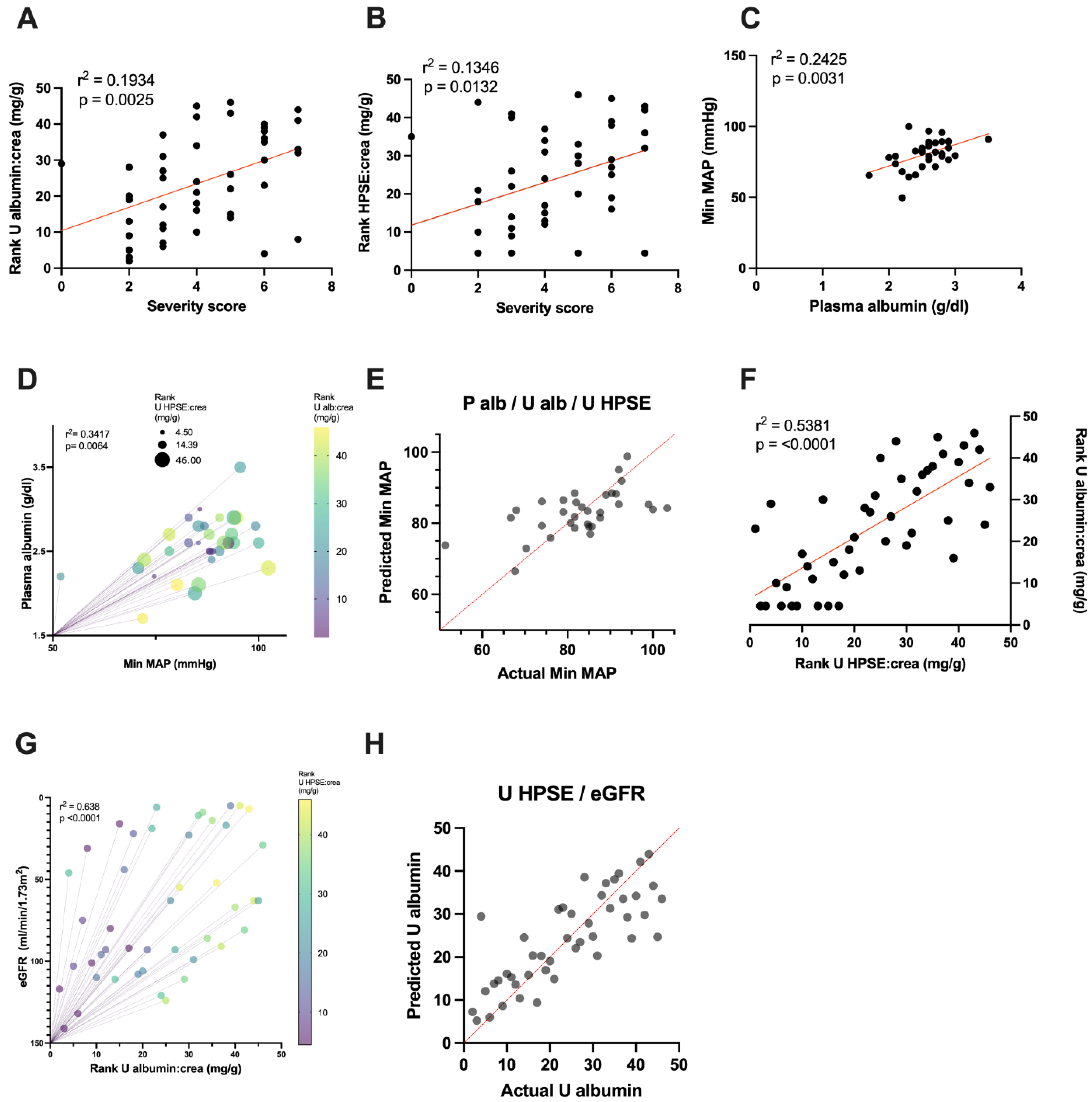
3.1. HPSE and Syndecan–1 Levels Are Increased in Urine of Acute PUUV–HFRS Patients
3.2. Urinary HPSE Levels Correlate with Albuminuria and Other Disease Severity Markers
3.3. Urinary HPSE Levels Possess Predictive Power over Disease Severity, Albuminuria and Hypotension
3.4. Upregulation of HPSE in Orthohantavirus–Infected Podocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef]
- Hepojoki, J.; Vaheri, A.; Strandin, T. The fundamental role of endothelial cells in hantavirus pathogenesis. Front Microbiol. 2014, 22, 727. [Google Scholar] [CrossRef] [Green Version]
- Mustonen, J.; Outinen, T.; Laine, O.; Pörsti, I.; Vaheri, A.; Mäkelä, S. Kidney disease in Puumala hantavirus infection. Infect. Dis. 2017, 49, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Ala–Houhala, I.; Koskinen, M.; Ahola, T.; Harmoinen, A.; Kouri, T.; Laurila, K.; Mustonen, J.; Pasternack, A. Increased glomerular permeability in patients with nephropathia epidemica caused by Puumala hantavirus. Nephrol. Dial. Transplant. 2002, 17, 246–252. [Google Scholar] [CrossRef]
- Mantula, P.; Tietäväinen, J.; Clement, J.; Niemelä, O.; Pörsti, I.; Vaheri, A.; Mustonen, J.; Mäkelä, S.; Outinen, T. Flash–Like Albuminuria in Acute Kidney Injury Caused by Puumala Hantavirus Infection. Pathogens 2020, 9, 615. [Google Scholar] [CrossRef]
- Mantula, P.S.; Outinen, T.K.; Clement, J.P.G.; Huhtala, H.S.A.; Pörsti, I.H.; Vaheri, A.; Mustonen, J.T.; Mäkelä, S.M. Glomerular Proteinuria Predicts the Severity of Acute Kidney Injury in Puumala Hantavirus–Induced Tubulointerstitial Nephritis. Nephron 2017, 136, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Boehlke, C.; Hartleben, B.; Huber, T.B.; Hopfer, H.; Walz, G.; Neumann–Haefelin, E. Hantavirus infection with severe proteinuria and podocyte foot–process effacement. Am. J. Kidney Dis. 2014, 64, 452–456. [Google Scholar] [CrossRef]
- Outinen, T.K.; Mäkelä, S.; Clement, J.; Paakkala, A.; Pörsti, I.; Mustonen, J. Community Acquired Severe Acute Kidney Injury Caused by Hantavirus–Induced Hemorrhagic Fever with Renal Syndrome Has a Favorable Outcome. Nephron 2015, 130, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Breshears, M.A.; Anthony, W.; Confer, A. Pathologic Basis of Veterinary Disease, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Rostgaard, J.; Qvortrup, K. Sieve plugs in fenestrae of glomerular capillaries—Site of the filtration barrier? Cells Tissues Organs 2002, 170, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Curry, F.E.; Adamson, R.H. Endothelial glycocalyx: Permeability barrier and mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Avasthi, P.S.; Koshy, V. The anionic matrix at the rat glomerular endothelial surface. Anat. Rec. 1988, 220, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Kerjaschki, D.; Sharkey, D.J.; Farquhar, M.G. Identification and characterization of podocalyxin—The major sialoprotein of the renal glomerular epithelial cell. J. Cell. Biol. 1984, 98, 1591–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ina, K.; Kitamura, H.; Nakamura, M.; Ono, J.; Takaki, R. Loss of sulfated carbohydrate from the glomerular podocyte as a cause of albuminuria in experimental diabetic rats: Ultrastructural histochemical study. J. Diabet. Complications. 1991, 5, 173–175. [Google Scholar] [CrossRef]
- Menzel, S.; Moeller, M.J. Role of the podocyte in proteinuria. Pediatr. Nephrol. 2011, 26, 1775–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krautkrämer, E.; Zeier, M.; Plyusnin, A. Hantavirus infection: An emerging infectious disease causing acute renal failure. Kidney Int. 2013, 83, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krautkrämer, E.; Grouls, S.; Stein, N.; Reiser, J.; Zeier, M. Pathogenic old world hantaviruses infect renal glomerular and tubular cells and induce disassembling of cell–to–cell contacts. J. Virol. 2011, 85, 9811–9823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusshag, C.; Stütz, A.; Hägele, S.; Speer, C.; Kälble, F.; Eckert, C.; Brenner, T.; Weigand, M.A.; Morath, C.; Reiser, J.; et al. Glomerular filtration barrier dysfunction in a self–limiting, RNA virus–induced glomerulopathy resembles findings in idiopathic nephrotic syndromes. Sci. Rep. 2020, 10, 19117. [Google Scholar] [CrossRef]
- Cosgriff, T.M. Mechanisms of disease in Hantavirus infection: Pathophysiology of hemorrhagic fever with renal syndrome. Rev. Infect. Dis. 1991, 13, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Gelberg, H.; Healy, L.; Whiteley, H.; Miller, L.A.; Vimr, E. In vivo enzymatic removal of alpha 2––>6–linked sialic acid from the glomerular filtration barrier results in podocyte charge alteration and glomerular injury. Lab. Invest. 1996, 74, 907–920. [Google Scholar] [PubMed]
- Jeansson, M.; Haraldsson, B. Glomerular size and charge selectivity in the mouse after exposure to glucosaminoglycan–degrading enzymes. J. Am. Soc. Nephrol. 2003, 14, 1756–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeansson, M.; Haraldsson, B. Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am. J. Physiol. Renal. Physiol. 2006, 290, F111–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwese, M.C.; Broekhuizen, L.N.; Kuikhoven, M.; Heeneman, S.; Lutgens, E.; Gijbels, M.J.; Nieuwdorp, M.; Peutz, C.J.; Stroes, E.S.; Vink, H.; et al. Endothelial surface layer degradation by chronic hyaluronidase infusion induces proteinuria in apolipoprotein E–deficient mice. PLoS ONE 2010, 5, e14262. [Google Scholar] [CrossRef] [Green Version]
- Dane, M.J.; van den Berg, B.M.; Avramut, M.C.; Faas, F.G.; van der Vlag, J.; Rops, A.L.; Ravelli, R.B.; Koster, B.J.; van Zonneveld, A.J.; Vink, H.; et al. Glomerular endothelial surface layer acts as a barrier against albumin filtration. Am. J. Pathol. 2013, 182, 1532–1540. [Google Scholar] [CrossRef]
- Kanwar, Y.S.; Farquhar, M.G. Presence of heparan sulfate in the glomerular basement membrane. Proc. Natl. Acad. Sci. USA 1979, 76, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, Y.S.; Farquhar, M.G. Isolation of glycosaminoglycans (heparan sulfate) from glomerular basement membranes. Proc. Natl. Acad. Sci. USA 1979, 76, 4493–4497. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, Y.S.; Linker, A.; Farquhar, M.G. Increased permeability of the glomerular basement membrane to ferritin after removal of glycosaminoglycans (heparan sulfate) by enzyme digestion. J. Cell Biol. 1980, 86, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, Y.S.; Jakubowski, M.L.; Rosenzweig, L.J. Distribution of sulfated glycosaminoglycans in the glomerular basement membrane and mesangial matrix. Eur. J. Cell. Biol. 1983, 31, 290–295. [Google Scholar]
- Thakkar, N.; Yadavalli, T.; Jaishankar, D.; Shukla, D. Emerging Roles of Heparanase in Viral Pathogenesis. Pathogens 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeansson, M.; Björck, K.; Tenstad, O.; Haraldsson, B. Adriamycin alters glomerular endothelium to induce proteinuria. J. Am. Soc. Nephrol. 2009, 20, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, E.; Cardenas, J.C.; Baimukanova, G.; Usadi, B.; Bruhn, R.; Pati, S.; Ostrowski, S.R.; Johansson, P.I.; Holcomb, J.B.; Wade, C.E. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J. Transl. Med. 2015, 13, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehm, M.; Bruegger, D.; Christ, F.; Conzen, P.; Thiel, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Reichart, B.; et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation 2007, 116, 1896–1906. [Google Scholar] [CrossRef] [Green Version]
- Rangarajan, S.; Richter, J.R.; Richter, R.P.; Bandari, S.K.; Tripathi, K.; Vlodavsky, I.; Sanderson, R.D. Heparanase–enhanced Shedding of Syndecan–1 and Its Role in Driving Disease Pathogenesis and Progression. J. Histochem. Cytochem. 2020, 68, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.H.; Aquino, R.S.; Park, P.W. Molecular functions of syndecan–1 in disease. Matrix Biol. 2012, 31, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly–Andersen, A.M.; Thunberg, T.; Ahlm, C. Endothelial activation and repair during hantavirus infection: Association with disease outcome. Open Forum Infect. Dis. 2014, 1, ofu027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. CKD–EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Buijsers, B.; Yanginlar, C.; de Nooijer, A.; Grondman, I.; Maciej–Hulme, M.L.; Jonkman, I.; Janssen, N.A.F.; Rother, N.; de Graaf, M.; Pickkers, P.; et al. Increased Plasma Heparanase Activity in COVID–19 Patients. Front. Immunol. 2020, 11, 575047. [Google Scholar] [CrossRef]
- Libraty, D.H.; Mäkelä, S.; Vlk, J.; Hurme, M.; Vaheri, A.; Ennis, F.A.; Mustonen, J. The degree of leukocytosis and urine GATA–3 mRNA levels are risk factors for severe acute kidney injury in Puumala virus nephropathia epidemica. PLoS ONE 2012, 7, e35402. [Google Scholar]
- Strandin, T.; Mäkelä, S.; Mustonen, J.; Vaheri, A. Neutrophil Activation in Acute Hemorrhagic Fever with Renal Syndrome Is Mediated by Hantavirus–Infected Microvascular Endothelial Cells. Front. Immunol. 2018, 9, 2098. [Google Scholar] [CrossRef] [Green Version]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real–time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- van den Hoven, M.J.; Waanders, F.; Rops, A.L.; Kramer, A.B.; van Goor, H.; Berden, J.H.; Navis, G.; van der Vlag, J. Regulation of glomerular heparanase expression by aldosterone, angiotensin II and reactive oxygen species. Nephrol. Dial. Transplant. 2009, 24, 2637–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabelink, T.J.; van den Berg, B.M.; Garsen, M.; Wang, G.; Elkin, M.; van der Vlag, J. Heparanase: Roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nat. Rev. Nephrol. 2017, 13, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, J.R.; Seiler, M.W. Pathophysiology of Tamm–Horsfall protein. Kidney Int. 1979, 16, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, M.H.; Makela, S.M.; Ala–Houhala, I.O.; Huhtala, H.S.; Koobi, T.; Vaheri, A.I.; Pasternack, A.I.; Porsti, I.H.; Mustonen, J.T. Tubular proteinuria and glomerular filtration 6 years after puumala hantavirus–induced acute interstitial nephritis. Nephron Clin. Pract. 2009, 112, c115–c120. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.H.; Mäkelä, S.M.; Ala–Houhala, I.O.; Huhtala, H.S.; Kööbi, T.; Vaheri, A.I.; Pasternack, A.I.; Pörsti, I.H.; Mustonen, J.T. Ten–year prognosis of Puumala hantavirus–induced acute interstitial nephritis. Kidney Int. 2006, 69, 2043–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Hoven, M.J.; Rops, A.L.; Vlodavsky, I.; Levidiotis, V.; Berden, J.H.; Van der Vlag, J. Heparanase in glomerular diseases. Kidney Int. 2007, 72, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymczak, M.; Kuźniar, J.; Klinger, M. The role of heparanase in diseases of the glomeruli. Arch. Immunol. Ther. Exp. 2010, 58, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Shafat, I.; Ilan, N.; Zoabi, S.; Vlodavsky, I.; Nakhoul, F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS ONE 2011, 6, e17312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hägele, S. Functional Consequences of Old World Hantavirus Infection in Human Renal Cells. Ph.D. Thesis, Heidelberg University, Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Hägele, S.; Müller, A.; Nusshag, C.; Reiser, J.; Zeier, M.; Krautkrämer, E. Virus– and cell type–specific effects in orthohantavirus infection. Virus Res. 2019, 260, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, D.; Vikramadithyan, R.; Yagyu, H.; Saxena, U.; Pillarisetti, S.; Goldberg, I. Inflammatory Cytokines and Fatty Acids Regulate Endothelial Cell Heparanase Expression. Biochemistry 2004, 43, 4971–4977. [Google Scholar] [CrossRef] [PubMed]
- Linderholm, M.; Ahlm, C.; Settergren, B.; Waage, A.; Tärnvik, A. Elevated Plasma Levels of Tumor Necrosis Factor (TNF)–α, Soluble TNF Receptors, Interleukin (IL)–6, and IL–10 in Patients with Hemorrhagic Fever with Renal Syndrome. J. Infec. Dis. 1996, 173, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Mahtal, N.; Lenoir, O.; Tharaux, P.L. Glomerular Endothelial Cell Crosstalk With Podocytes in Diabetic Kidney Disease. Front. Med. 2021, 8, 659013. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable (Unit) | Mean (SE; Range) |
|---|---|
| Number of patients | 56 |
| Hospital length of stay (days) | 5.81 (0.46; 2–25) |
| Age (years) | 41 (1.65; 22–73) |
| Male: Female ratio | 2.3:1 |
| Min eGFR (mL/min/1.73m2) | 63 (5.75; 5–141) |
| Max plasma creatinine (μmol/L) | 266.22 (35.61; 45–1071) |
| Urine albumin: creatinine ratio (mg/g) | 1012.12 (190.67; 2.9–5530.8) |
| Max urine HPSE: creatinine ratio (mg/g) | 0.22 (0.04; 0–1.17) |
| Min plasma albumin (g/dL) | 2.41 (0.08; 2.3–3.5) |
| Max plasma HPSE (ng/mL) | 4.54 (0.70; 0–20.23) |
| Max urine syndecan–1: creatinine ratio | 36.3 (9.5; 0–411.62) |
| Urine IL–6: creatinine ratio (pg/μg) | 0.21 (0.05; 0.005–1.30) |
| Plasma IL–6 (pg/mL) | 128.58 (39.9; 10–785.8) |
| Urine IL–8: creatinine ratio (pg/μg) | 0.45 (0.18; 0.003–5.80) |
| Plasma IL–8 (pg/mL) | 41.26 (8.72; 10–234.86) |
| Urine IP–10: creatinine ratio (pg/μg) | 4.47 (0.70; 0.22–16.92) |
| Plasma IP–10 (pg/mL) | 4734.39 (764.17; 739.8–18224) |
| Urine MCP–1: creatinine ratio (pg/μg) | 7.64 (1.97; 0.01–63.64) |
| Plasma MCP–1 (pg/mL) | 373.97 (120.78; 23.5–3636.78) |
| MPO (ng/mL) | 150.85 (9.5; 46–332.17) |
| Severity score | 4.04 (0.24; 0–7) |
| Min mean arterial pressure (mmHg) | 84.11 (1.42; 51.33–103.33) |
| Max CRP (mg/L) | 67.32 (5.11; 9.7–155.2) |
| Min thrombocytes (109/L) | 85.56 (5.66; 36–266) |
| Max leukocytes (109/L) | 10.62 (0.58; 4.1–25.5) |
| Dependent Variables | Type of Regression | Independent Variables | Estimate | Std. Error | 95% CI | t Value | p–Value of Variable | R2 | F– Statistic | p–Value of Model |
|---|---|---|---|---|---|---|---|---|---|---|
| Minimum mean arterial pressure | Simple | Plasma albumin | 15.594 | 4.715 | [5.488–24.7] | 3.440 | 0.0031 ** | 0.2425 | 10.25 | 0.0031 ** |
| Multiple | Plasma albumin | 15.4735 | 4.8731 | [5.507–25.44] | 3.175 | 0.0035 ** | 0.3417 | 5.017 | 0.0064 ** | |
| Urinary albumin: creatinine ratio | −0.1508 | 0.1774 | [−0.5136–0.2121] | −0.85 | 0.0724 | |||||
| Urinary HPSE: creatinine ratio | 0.3703 | 0.1854 | [−0.0088–0.7495] | 1.998 | 0.0502 | |||||
| Severity score | Simple | Urinary albumin: creatinine ratio | 3.254 | 1.014 | [1.210–5.299] | 3.211 | 0.0025 ** | 0.1934 | 10.31 | 0.0025 ** |
| Simple | Urinary HPSE: creatinine ratio | 2.799 | 1.082 | [0.6167–4.982] | 2.586 | 0.0132 * | 0.1346 | 6.690 | 0.0132 * | |
| Urinary albumin: creatinine ratio | Simple | Urinary HPSE: creatinine ratio | 0.7355 | 0.1027 | [0.5257–0.9376] | 0.1027 | <0.0001 *** | 0.5276 | 51.26 | <0.0001 *** |
| Multiple | Urinary HPSE: creatinine ratio | 0.6587 | 0.0941 | [0.4687–0.8486] | 6.998 | <0.0001 *** | 0.621 | 37.02 | <0.0001 *** | |
| eGFR | −0.0848 | 0.0308 | [−0.1469–−0.0227] | −2.756 | 0.0086 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera, L.E.; Schmotz, C.; Saleem, M.A.; Lehtonen, S.; Vapalahti, O.; Vaheri, A.; Mäkelä, S.; Mustonen, J.; Strandin, T. Increased Heparanase Levels in Urine during Acute Puumala Orthohantavirus Infection Are Associated with Disease Severity. Viruses 2022, 14, 450. https://0-doi-org.brum.beds.ac.uk/10.3390/v14030450
Cabrera LE, Schmotz C, Saleem MA, Lehtonen S, Vapalahti O, Vaheri A, Mäkelä S, Mustonen J, Strandin T. Increased Heparanase Levels in Urine during Acute Puumala Orthohantavirus Infection Are Associated with Disease Severity. Viruses. 2022; 14(3):450. https://0-doi-org.brum.beds.ac.uk/10.3390/v14030450
Chicago/Turabian StyleCabrera, Luz E., Constanze Schmotz, Moin A. Saleem, Sanna Lehtonen, Olli Vapalahti, Antti Vaheri, Satu Mäkelä, Jukka Mustonen, and Tomas Strandin. 2022. "Increased Heparanase Levels in Urine during Acute Puumala Orthohantavirus Infection Are Associated with Disease Severity" Viruses 14, no. 3: 450. https://0-doi-org.brum.beds.ac.uk/10.3390/v14030450