1. Introduction
Newly assembled herpesvirus capsids translocate from the nucleus to the cytoplasm in a complicated process known as nuclear egress. Nuclear egress includes four distinct steps: (1) migration of capsids from the nuclear interior, where viral genome replication and encapsidation occur within discrete replication compartments (RCs), to the nuclear rim; (2) disruption of the nuclear lamina providing access to the inner nuclear membrane (INM): (3) budding of capsids through the INM (primary envelopment); and (4) de-envelopment at the outer nuclear membrane (reviewed in [
1,
2,
3,
4]).
Step 1 of this process—migration from the nuclear interior to the nuclear rim—is poorly understood. In one model, as initially proposed for an alphaherpesvirus, herpes simplex virus 1 (HSV-1) [
5] capsids move from the nuclear interior towards the nuclear rim by actomyosin-directed transport. For a betaherpesvirus, human cytomegalovirus (HCMV), this model is supported for at least some intranuclear capsid migration by data showing that the virus induces the formation of actin filaments in the nucleus that associate with capsids and myosin Va, that treatment with an actin-depolymerizing drug disrupts these filaments and impairs migration to the nuclear rim and nuclear egress, and that siRNA and a nuclear localized dominant negative mutant that antagonize myosin Va also impair these processes [
6,
7]. However, how the capsid would interact with the actomyosin machinery to be transported from the nuclear interior to the nuclear rim remains unclear.
Another poorly understood aspect of nuclear egress is the relationship between this first step of nuclear egress and the viral nuclear egress complex (NEC). The NEC consists of two virus-encoded subunits, one that is anchored in the INM, and the other that binds to its nucleoplasmic face. For HCMV, the INM-anchored subunit is UL50 and the nucleoplasmic subunit is UL53. Both subunits are essential for viral replication and nuclear egress [
8,
9,
10]. The HCMV NEC recruits the viral protein kinase, UL97, to the nuclear rim for phosphorylation and thus disruption of the nuclear lamina [
10,
11]. Based on work with other herpesvirus NECs, the HCMV NEC also likely orchestrates capsid budding during primary envelopment (reviewed in [
1,
2,
3,
4]). However, there is evidence that nucleoplasmic subunits of certain herpesvirus NECs participate in processes upstream of events at the nuclear rim, such as DNA packaging, and capsid migration to the nuclear periphery [
12,
13,
14,
15,
16]. Moreover, homologs of UL53, such as the HSV-1 and pseudorabies virus nucleoplasmic NEC subunit, UL31, have been found to associate with intranuclear capsid proteins [
16,
17,
18], and recently, some evidence that HCMV UL53 associates with intranuclear capsids was provided [
19]. We were intrigued by these reports, as they raised the possibility that HCMV UL53 might interact with both capsids and the actomyosin machinery to mediate migration of HCMV capsids to the nuclear rim.
This study began with a proteomics investigation to identify candidate proteins that interact with UL53. This identified possible associations of UL53 with myosin Va and with capsid proteins, and we found several lines of evidence for modest associations of these components that were consistent with the hypothesis that UL53 might serve as a bridge between the actomyosin machinery and capsids during their migration to the nuclear rim. We then generated complementing cell lines to allow us to derive stocks of viral mutants with sufficiently high titers to allow us to test whether UL53 was required for associations of myosin Va with capsids, and for capsid migration towards the nuclear rim.
2. Materials and Methods
2.1. Cells and Viruses
Human foreskin fibroblasts (HFF) cells (ATCC, CRL-1684) and human embryonic kidney (293T) cells (ATCC (Manassas, VA, USA), CRL-11268) were propagated in DMEM containing 10% fetal bovine serum (FBS). The HCMV laboratory strain AD169 was used in all experiments. AD169-RV encoding a FLAG-tagged version of UL53 (53-F), and bacterial artificial chromosomes (BACs) encoding this virus and UL53-null (53N), UL50-null (50N), and UL53-null rescue-derivative (53NR) viruses have been described previously [
10]. The production of HFFs expressing UL50 or UL53 and infectious 50N, 53N, and 53NR viruses is described below. Viruses were propagated and titrated as described previously [
10,
20].
2.2. Mass-Spectrometry
Immunoprecipitation (IP) of UL53-FLAG from nuclear lysates of infected cells for mass spectrometry was carried out as described previously [
21]. Briefly, HFFs were infected with 53-F or WT HCMV (MOI 3) and cells were harvested at 72 h post-infection (hpi). Nuclear fractions were then isolated and subjected to α-FLAG IP. For mass spectrometry, eluates in Laemmli buffer were run on a 4–20% SDS-polyacrylamide gel. Extracted bands were submitted to the Taplin Mass-Spectrometry Facility (Harvard Medical School, Boston, MA, USA) for liquid chromatography-tandem mass-spectrometry (LC-MS/MS) analysis.
2.3. Immunoprecipitation
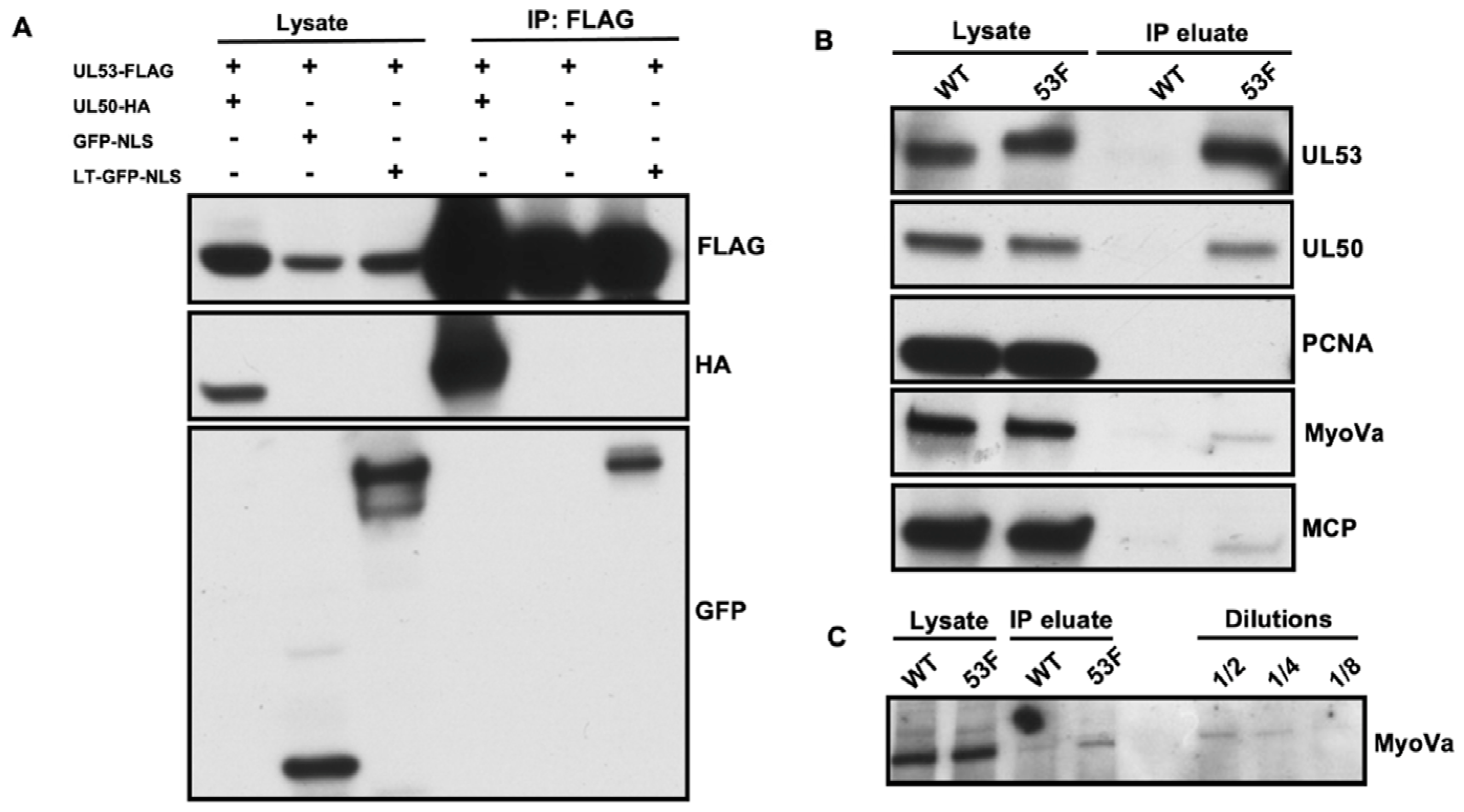
For immunoprecipitation (IP) of transfected cells, 5 × 10
6 293T cells/plate were seeded in 100 mm plates. Cells were then transfected with a pcDNA vector encoding a FLAG-tagged version of UL53 (UL53-FLAG) [
22] and either a pcDNA-based plasmid encoding an HA-tagged version of UL50 (UL50-HA) [
22] a lentiviral vector encoding green fluorescent protein with a nuclear localization sequence (GFP-NLS, [
7]), or a lentiviral vector encoding the long tail of myosin Va fused to GFP-NLS (LT-GFP-NLS, [
7]) (total of 10 μg DNA/plate; 1 μg/mL doxycycline was added to induce expression from the lentiviral vectors). At 48 h post-transfection, the cell monolayers were washed with Dulbecco’s phosphate-buffered saline (DPBS) and whole cell lysates were harvested in EBC buffer (50 mM Tris (pH 8.0), 150 mM NaCl, 0.5% NP-40) containing one Complete EDTA-free protease inhibitor tablet (Roche, Indianapolis, IN, USA) per 50 mL. For IP, 25 μL of EZ-View α-FLAG M2 affinity gel (Sigma, St. Louis, MO, USA) was added to lysates and rotated overnight at 4 °C. Resin was centrifuged and washed 4 times in 750 μL ice-cold EBC buffer by rotating for 20 min at 4 °C between washes. After the final spin, the resin pellet was mixed with 25 μL of EBC buffer, and protein was eluted from resin by incubation with 50 μL of 2x Laemmli buffer at 95 °C for 5 min and analyzed by Western blot as described below.
For IP of UL53-FLAG from infected cells, 2.5 × 106 HFFs were infected with either 53-F or WT HCMV (MOI 1). At 72 hpi, cells were harvested, and nuclei were isolated and lysed using a Nuclear Complex Co-IP Kit (Active Motif, Carlsbad, CA, USA). The nuclear lysate was mixed with 1 mL EBC buffer containing one Complete EDTA-free protease inhibitor tablet (Roche) per 50 mL and precleared with 100 μL of mouse IgG-agarose (Sigma) by rotating at 4 °C for 5 h. For IP, 40 μL of EZ-View α-FLAG M2 affinity gel (Sigma) was added to precleared lysate and rotated overnight at 4 °C. Resin was centrifuged and washed 4 times in 750 μL ice-cold EBC buffer by rotating for 20 min at 4 °C between washes. After the final spin, the resin pellet was mixed with 40 μL of EBC buffer, and protein was eluted from resin by incubation with 80 μL of 2x Laemmli buffer at 95 °C for 5 min and analyzed by Western blot as described below.
2.4. Western Blotting
For Western blotting of IPs, lysates and eluates in Laemmli buffer were separated on a 4–20% SDS-polyacrylamide gel (Bio-Rad, Hercules, CA, USA). Proteins were then transferred onto a PVDF membrane, blocked with 5% milk in DPBS-T (DPBS with 0.5% Tween-20), and probed with primary antibodies (see below for sources and dilutions) overnight at 4 °C with rocking. Membranes were washed 3x with DPBS-T for 10 min at room temperature (RT) with rocking. Membranes were then incubated with TrueBlot secondary antibodies conjugated to horseradish peroxidase (HRP) (Rockland, Limerick, PA, USA) at 1:1000 for 1 h at RT with rocking, followed by washing. Finally, Pierce chemiluminescence solution (ThermoFisher, Waltham, MA, USA) was added to membranes and signal was detected with film.
For all other Western blotting, cells were harvested by washing with DPBS followed by the addition of 2x Laemmli buffer with protease inhibitors (ThermoFisher) directly to the monolayer. Lysates were scraped off the plate, boiled at 95 °C for 5 min, and processed as described above.
Primary antibody dilutions were as follows: rabbit α-myosin Va (Cell Signaling Technology, Danvers, MA, USA, #3402), 1:1000; mouse α-β actin (Sigma A5441), 1:5000; mouse α-FLAG M2 (Sigma F1804), 1:100; rabbit α-UL53 [
10], 1:500; rabbit α-UL50 [
10], 1:500; rabbit α-PCNA (Abcam, Cambridge, UK, ab18197), 1:700; mouse α-MCP (a kind gift from William Britt, University of Alabama, Birmingham, AL, USA), 1:1000; rabbit α-GFP (ThermoFisher A11122), 1:1000.
2.5. Immunofluorescence Analysis
For immunofluorescence, 1 × 105 HFFs/well were seeded on glass coverslips in a 24-well plate followed by either mock infection or infection with WT, 53-F, 50N/53F HCMV (MOI 1) as indicated in the text. At the time-points indicated, cells were fixed at RT in 3.7% formaldehyde/DPBS. Cells were then permeabilized at RT in 0.1% Triton X-100/DPBS, washed 3x with DPBS, and blocked overnight in a mixture of 1% bovine serum albumin (Sigma) and 5% human serum (Sigma) in DPBS. The following antibodies and dilutions were used for primary staining: rabbit α-myosin Va (Abcam, ab11094), 1:50; mouse α-FLAG M2 (Sigma F1804), 1:500; mouse α-MCP (a kind gift from William Britt, University of Alabama, Birmingham, AL, USA), 1:250. Antibodies were diluted in a mixture of 1% BSA/5% human serum in DPBS and added to coverslips for 1 h at RT with rocking. Primary antibodies were removed and coverslips washed 3x with DPBS for 5 min with rocking at RT. The staining procedure was repeated with the appropriate fluorescently labeled Alexa-fluor secondary antibodies (ThermoFisher), and DAPI was applied in the last 10 min of the secondary antibody incubation. After the final washes, coverslips were mounted on glass slides using ProLong Anti-fade (ThermoFisher). Imaging was carried out at the Nikon Imaging Center (NIC) at Harvard Medical School using a Nikon Ti spinning-disk confocal laser microscope. Postacquisition image analysis was conducted using Metamorph and ImageJ software packages.
2.6. Immunoelectron Microscopy
Here, 2 × 105 HFFs/well were seeded in 12-well plates and infected with either WT or 53-F HCMV (MOI 1). At 72 hpi, cells were washed with DPBS, trypsinized, and harvested. The cell suspension was layered on top of a cushion of 4% paraformaldehyde and 0.1% glutaraldehyde in DPBS and pelleted for 3 min at 3000 rpm. The supernatant was carefully removed and fresh 4% paraformaldehyde and 0.1% glutaraldehyde were added. After fixation at RT for 2 h, the fixative was replaced with DPBS. Prior to freezing in liquid nitrogen the cell pellets were infiltrated with 2.3 M sucrose in DPBS (containing 0.2 M glycine to quench free aldehyde groups) for 15 min. Frozen samples were sectioned at −120 °C, the sections were transferred to formvar-carbon coated copper grids. Grids were floated on DPBS or stored on 2% gelatin dishes at 4 °C until immunogold labeling. The gold labeling was carried out at RT on a piece of parafilm. Antibodies and protein A gold were diluted in 1% BSA in PBS. The diluted primary antibody solution (α-myosin Va 1:10) was centrifuged 1 min at 14,000 rpm prior to labeling to avoid possible aggregates. Grids were floated on drops of 1% BSA for 10 min to block for unspecific labeling, transferred to 5 µL drops of primary antibody and incubated for 30 min. The grids were then washed in 4 drops of DPBS for a total of 15 min, transferred to 5 µL drops of 10 nm Protein A gold for 20 min, washed in 4 drops of DPBS for 15 min and 6 drops of double-distilled water. Contrasting/embedding of the labeled grids was carried out on ice in 0.3% uranyl acetate in 2% methyl cellulose for 10 min. Grids were picked up with metal loops (diameter slightly larger than the grid) and the excess liquid was removed by streaking on filter paper, leaving a thin coat of methylcellulose.
The grids were examined on a JEOL 1200EX electron microscope and images were recorded with an AMT 2k CCD camera. Labeled and unlabeled capsids were counted, and the percentage of capsids associated with at least one gold particle was calculated for each condition, and analyzed by Fisher’s exact test using GraphPad Prism Version 7 software, GraphPad Software, San Diego, CA, USA.
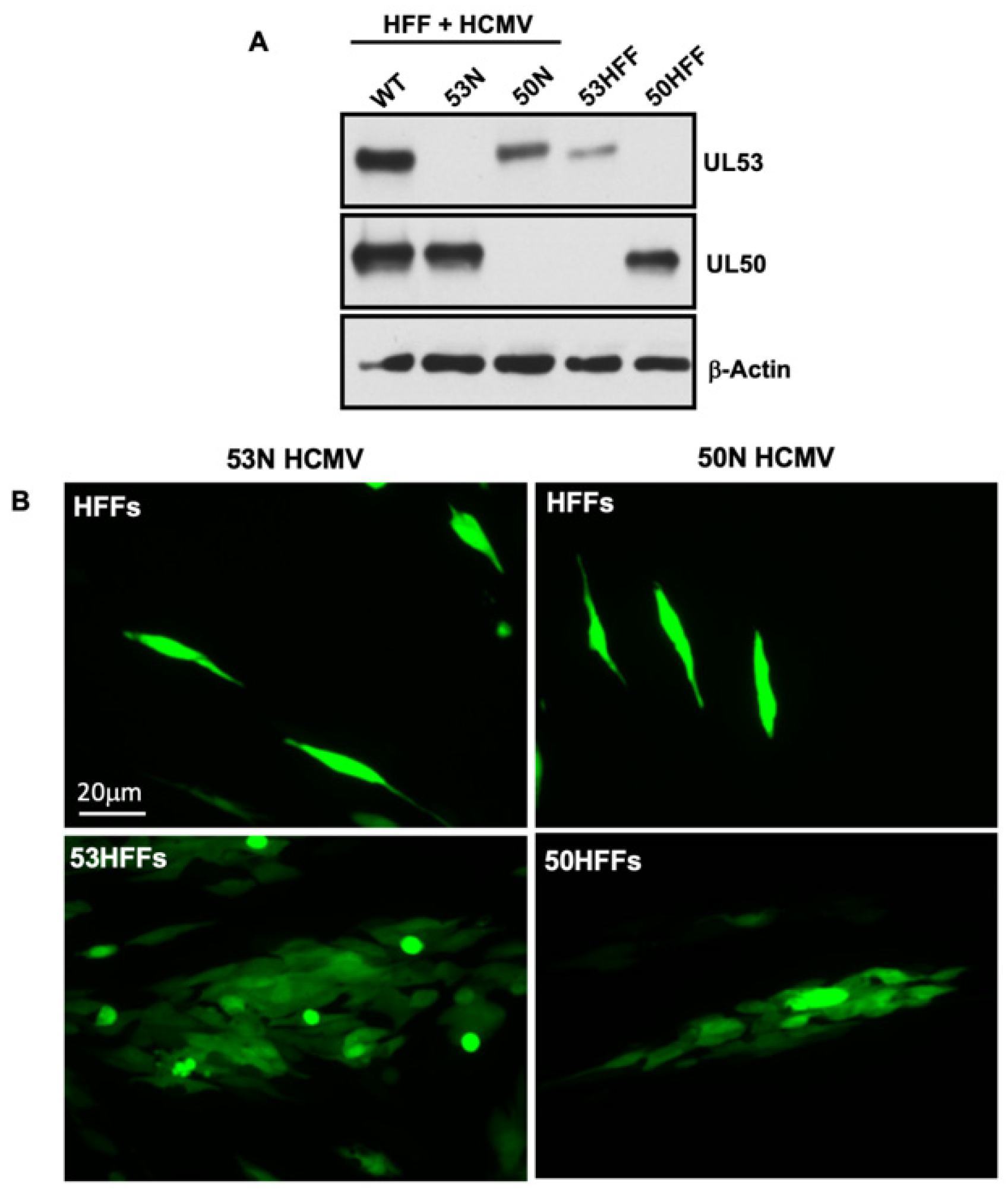
2.7. Generation of Infectious 53N and 50N Viruses
To generate HFFs stably expressing either UL53 or UL50, each viral gene was amplified from WT HCMV BAC DNA with the following primers: UL53, forward: 5′-TAAGCAGCGGCCGCATGTCTAGCGTGAGCGGC GTGCGCA-3′; UL53, reverse: 5′-TGCTTAGGATCCTCAAGGCGCACGAATGCTGTTGAGAAACAGCGG-3′; UL50, forward: 5′-TAAGCAGCGGCCGCATGGAGATGAACAAGG TTCTCCATC-3′; UL50, reverse: 5′-TGCTTAGGATCCTCAGTCGCGGTGTGCGGAGCGTGTCGGA-3′. Each PCR product was digested with Not1 and BamH1 restriction enzymes and cloned into the pLVX-eF1αlentiviral vector (generous gift from the late Gregory Pari, University of Nevada, Reno, NV, USA). Lentiviruses were produced following transfection of 293T with these pLVX-eIFα-based plasmids and used to transduce HFFs as described previously [
7]. The UL53 expressing cell lines were then electroporated with WT, 53N, or 53NR BACS, and the UL50 expressing cell line was electroporated with WT or 50N HCMV BAC DNA. After several weeks, viral supernatant was harvested and used for experiments as described in the text.
2.8. Transmission Electron Microscopy
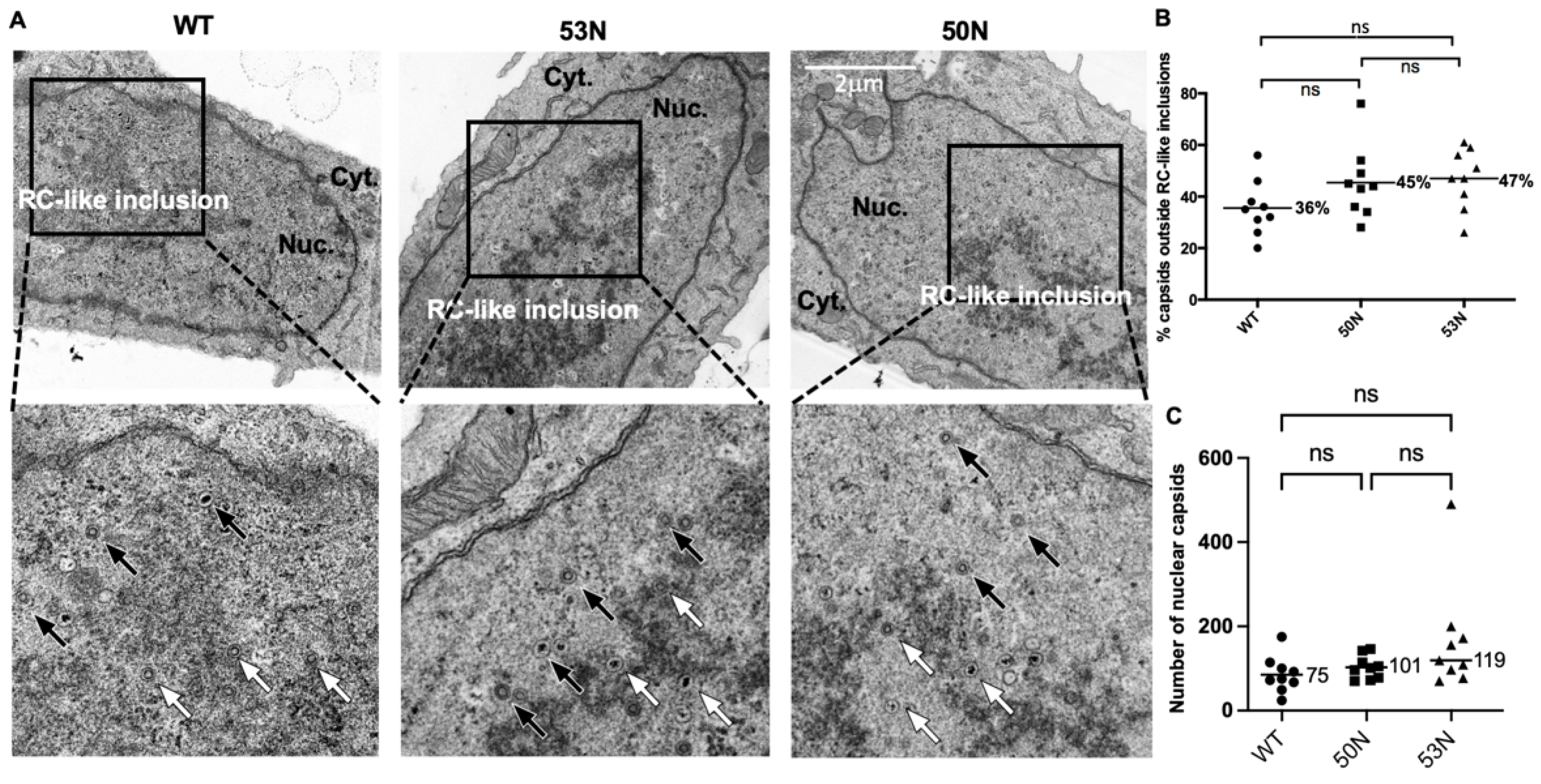
Transmission electron microscopy (TEM) was utilized to assess subnuclear capsid distribution by counting capsids in the nuclei either inside or outside of RC-like inclusions under the conditions described in the text using a TecnaiG² Spirit BioTWIN electron microscope equipped with an AMT 2k CCD camera. Processing for image acquisition was performed as described previously [
23]. Intranuclear capsid distributions were assessed by counting capsids within or outside of electron dense inclusions in the interior of the nucleoplasm that have been considered to be RCs (e.g., [
15]) in representative sections of whole nuclei [
6]. We term these RC-like inclusions. Data analyses for capsid distributions were performed as described previously [
23], using ordinary one-way ANOVA corrected for multiple comparisons using the Holm–Sidak test, while analyses of capsid counts were performed using the Kruskal–Wallis test and Dunn’s tests for multiple comparisons. All statistical analyses used GraphPad Prism version 7 (capsid distributions) and version 9.3.1 for Mac (capsid counts).
2.9. Correlative Light Electron Microscopy
Correlative light electron microscopy (CLEM) was conducted as described previously [
10]. Briefly, HFFs were electroporated with either 53N or 53NR BAC DNA. The following day, cells were seeded onto gridded glass bottom dishes, and on day 7 or 8 postelectroporation they were fixed, imaged with fluorescence and phase microscopy to visualize the electroporated cells and the grid, and processed for EM. GFP-positive cells were identified by their grid coordinates, excised, and remounted for serial sectioning. Imaging was carried out using a TecnaiG
2 Spirit BioTWIN microscope. Representative whole-cell sections from three GFP-positive cells containing capsids were analyzed for each condition. Fisher’s exact test was applied to data using GraphPad Prism software version 7.
4. Discussion
How nascent herpesvirus capsids migrate from RCs in the nuclear interior to the periphery during nuclear egress is poorly understood. It is particularly unclear whether the NEC or its subunits, which are crucial for later steps of nuclear egress, play a role in this migration. We performed a mass spectrometry study to look for proteins that associate directly or indirectly with the nucleoplasmic subunit of the HCMV NEC, UL53, and identified myosin Va and capsid proteins as candidate UL53-interacting proteins. Follow-up studies provided evidence to validate these associations, although they were rather modest. Our results together with results reported previously in various herpesvirus systems [
5,
6,
7,
12,
13,
14,
15,
16,
19], led to the hypothesis that UL53 might serve as a bridge between capsids and nuclear actomyosin machinery. To test this role for UL53 and, more generally, a role in migration from the nuclear interior to the nuclear rim, we generated stocks of
UL50- and
UL53-null viruses using newly derived complementing cells. Experiments using these mutant viruses led to the conclusion that UL53 is neither important for associations between capsids and myosin Va nor for capsid localization away from RC-like inclusions in the nuclear interior. We discuss each of these findings below.
Our mass spectrometry, co-IP, IFA, and immunoEM results suggested associations among UL53, myosin Va, and major capsid proteins or assembled capsids in infected nuclei. By IFA and immunoEM, during WT infection, myosin Va and MCP or capsid associations were found largely within RCs or RC-like inclusions in the nuclear interior, while most UL53 could be found at the nuclear rim (Ref.[
7] and this study). However, even during WT infection, a subpopulation of UL53 was also evident in the nuclear interior where it colocalized with myosin Va and MCP (as shown by IFA) or capsids (as shown by immunoEM—the IFA studies do not distinguish between capsids and unassembled MCP or partially assembled particles). This is consistent with reports that the HSV-1 homolog of UL53 (HSV-1 UL31), co-localizes with MCP hexons and associates with the portal vertex of capsids in the nucleoplasm [
16,
17]. The results are also consistent with a report that HCMV UL53 could be found in co-IP using antibodies against smallest capsid protein, and on intranuclear capsids by immunoEM [
19]. However, the capsids shown in that report were close to the nuclear envelope rather than in RC-like inclusions. Regardless, the relatively small amount of nucleoplasmic UL53 involved in associations with myosin Va and capsids likely reflects the small proportion that is not bound to UL50 at the INM under steady state conditions. Consistent with that surmise, UL53 primarily localizes to RCs during 50N infection, where we detected more obvious colocalization of UL53 with myosin Va or MCP.
Thus, we speculate that, ordinarily, some UL53 and myosin Va associate initially with capsids in RCs, and that subsequent movement to the nuclear periphery and primary envelopment occur relatively quickly and are thus not easily detected in fixed cells. For interactions between UL53 and capsids, there is considerable evidence for association of UL53 homologs of other viruses with various capsid proteins, particularly HSV-1 UL31 with HSV-1 UL25, or inner tegument proteins [
17,
18,
27,
28,
29,
30]. A recent study made the exciting finding that in HSV-1, UL25, interacts with the NEC and promotes formation of pentagonal arrays, which are posited to anchor capsids to the NEC and promote NEC curvature [
30]. The HCMV homolog of HSV-1 UL25, UL77, was not one of the proteins detected in our MS study, although we did detect MCP, the portal protein UL104, and several other components of intranuclear capsids including the terminase subunits, UL56 and UL89, and the inner tegument protein, UL32 (pp150), as well as TRS1, which has been reported to abet capsid assembly (
Table S1 in Supplementary Materials). Our failure to detect UL77 may reflect limitations of the mass spectrometric approach, our use of soluble nuclear lysates, or differences in the HCMV and HSV-1 systems.
In contrast with the literature on associations of UL53 homologs with capsids and/or capsid proteins, there is little evidence regarding how myosin Va associates with these and with UL53. Such associations could be direct or indirect. Given these associations and previous studies indicating a role for UL53 homologs and myosin Va in intranuclear distribution of capsids [
7,
15,
16] the hypothesis that UL53 might serve as an adaptor protein, bridging capsids and myosin Va, was attractive. However, deletion of UL53 did not discernibly affect the association between capsids and myosin Va. Thus, the details of UL53 interactions with myosin Va and nucleoplasmic capsids remain unclear.
Our finding that a population of UL53 localizes to the nucleoplasm was consistent with reports that UL53 homologs participate in events upstream of primary envelopment. For example, roles for UL53 homologs in viral DNA packaging (or stabilization of filled capsids) have been described for α-, β-, and γ-herpesviruses [
12,
13,
14,
15]. Furthermore, UL53 homologs have been suggested to facilitate capsid migration to the nuclear periphery. In one paper, it was stated in the Discussion that in cells replicating Epstein–Barr virus genomes lacking its
UL53 homolog, capsids were homogeneously distributed throughout the nucleus rather than being mostly aligned along the nuclear membrane [
13]. In a second paper, in one of the figures, capsids derived from MCMV expressing a dominant-negative version of its UL53 homolog, M53, clustered in the nuclear interior away from the nuclear rim [
15]. These authors went on to suggest that impaired viral DNA packaging observed with this mutant (and with another dominant-negative
M53 mutant [
14]) causes capsids to stall at packaging sites and that M53 mediates MCMV capsid localization to the nuclear periphery [
15]. Subsequently, it was shown in an interesting paper [
16] that an HSV-1 mutant, in which two basic patches of the N-terminal segment of its UL53 homolog, UL31, were made less basic, is defective for nuclear egress. IFA of UL31 and capsid proteins, particularly in cells transfected with a BAC containing the mutant genome, led to the conclusion that the N-terminal segment is required to direct migration of nucleocapsids to sites of primary envelopment at the nuclear periphery [
16].
While our initial CLEM results, using cells into which BACs containing the null mutant genome were introduced, raised the possibility that UL53 is important for capsid localization towards the nuclear periphery, subsequent analysis of a larger number of cells that were directly and synchronously infected by null mutant viruses indicated that deletion of
UL53 (or
UL50) did not result in altered intranuclear localization of capsids. (We also observed that copious DNA-filled C capsids formed in the absence of UL53, as we had previously [
10], indicating that UL53 is not essential for viral DNA packaging or for stabilization of C capsids.) It is possible that UL53 differs from its EBV, MCMV, and HSV-1 homologs in its ability to promote capsid migration to the nuclear rim. It is also possible that certain differences in results among the systems might reflect differences in how experiments were performed such as differences in preparation of infected cells (e.g., transfection vs. infection), mutants used (e.g., null vs. ones retaining certain activities), and whether and how intranuclear distributions of capsids were quantified.
What role does UL53 interactions with nucleoplasmic capsids play? One possibility is that these interactions are transient and/or infrequent and play no role. A second speculative possibility is that any UL53 that associates with capsids at the RC and migrates with them to the nuclear rim is then able to dock those capsids to any unoccupied UL50 at the INM for subsequent primary envelopment. Such a role would not depend on whether capsids migrate by the nuclear actomyosin machinery [
5] or by random diffusion [
31,
32]. Additional studies are required to address these possibilities.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






