
SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic

 , , , , , , , , ,
, , , , , , , , ,  ,
,  , , and
, , and 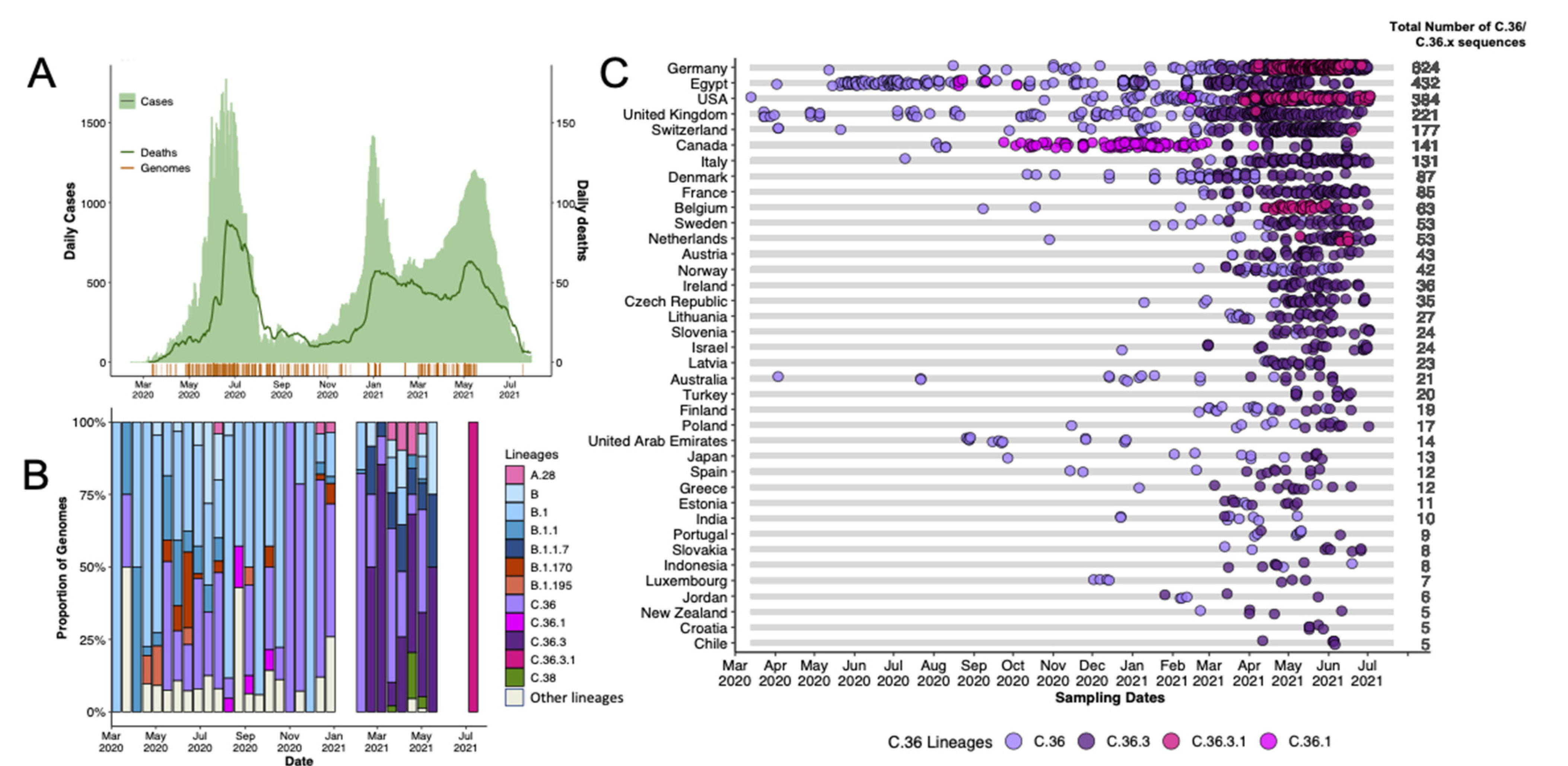{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Epidemiological Data
2.3. qPCR analyzed Samples
2.4. Mutation Screening Assays
2.5. SARS-CoV-2 Whole-Genome Sequencing
2.6. Genomic Analysis and Strain Typing
2.7. Phylogenetic Analysis
3. Results
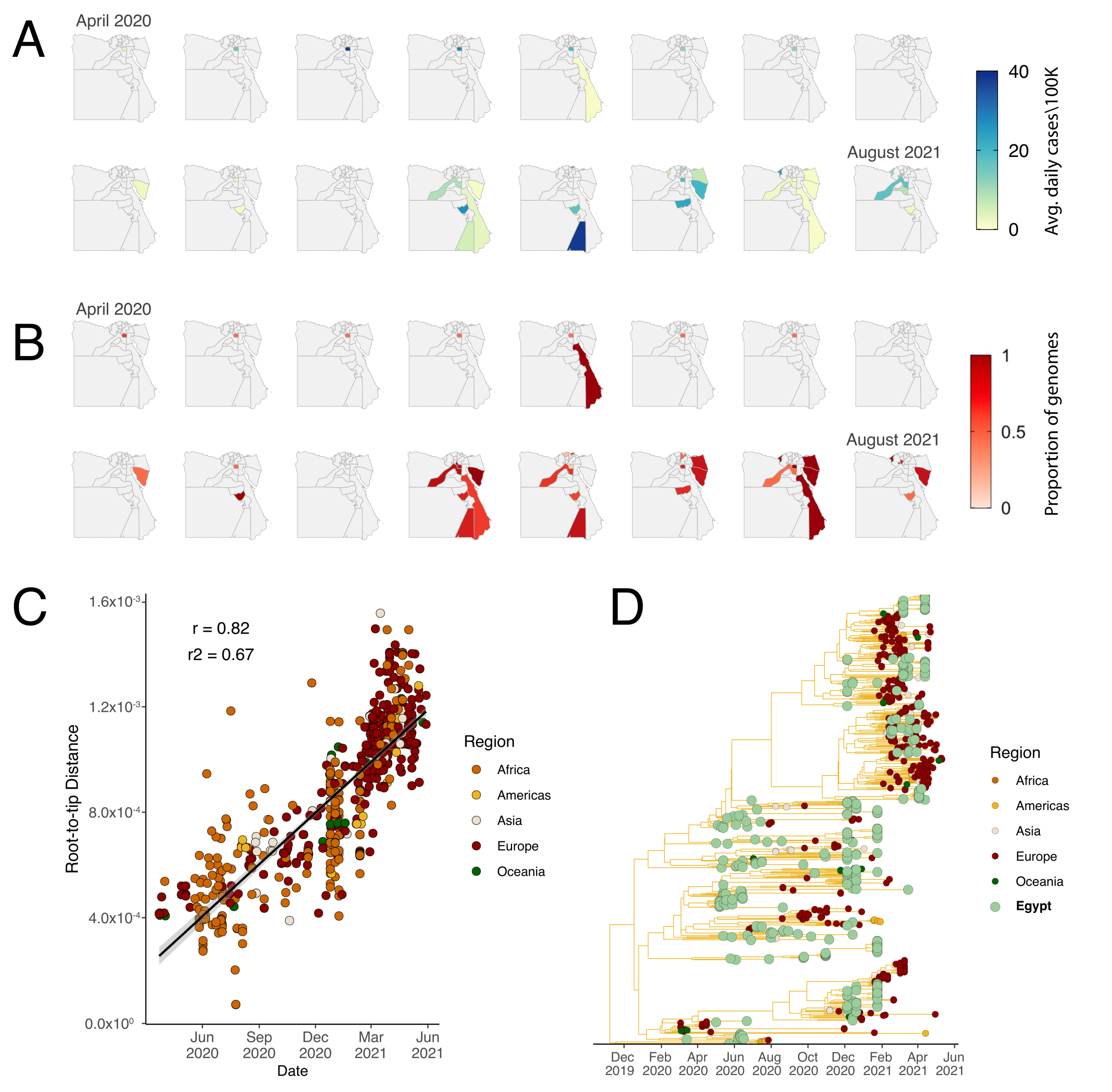
3.1. Genomic Epidemiology of SARS-CoV-2 in Egypt

3.2. Phylogenetic Analysis and Lineage Dynamics

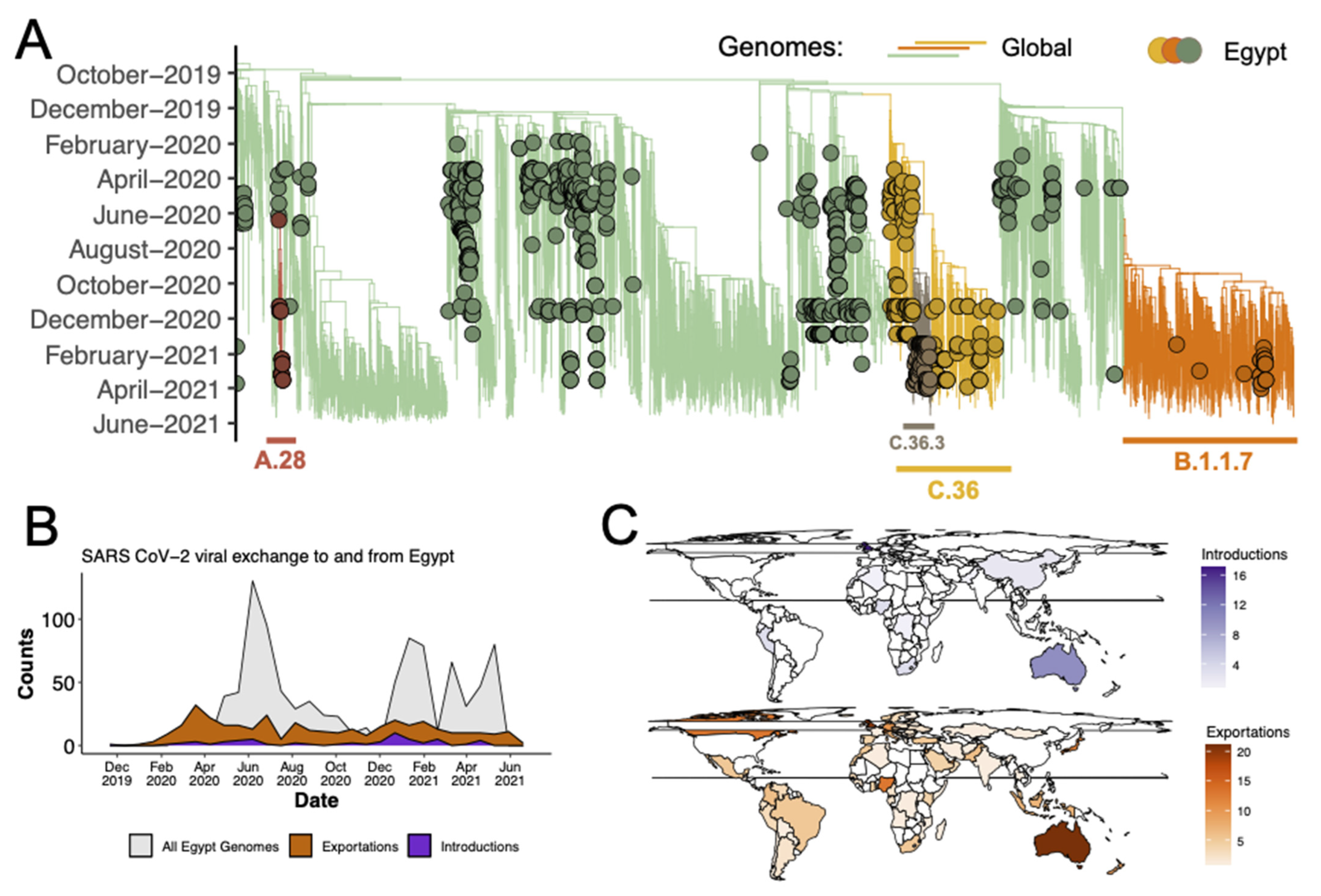
3.3. Introduction of the C.36 lineage in Egypt
3.4. Evolution and Spread of C.36 Sub-Lineages

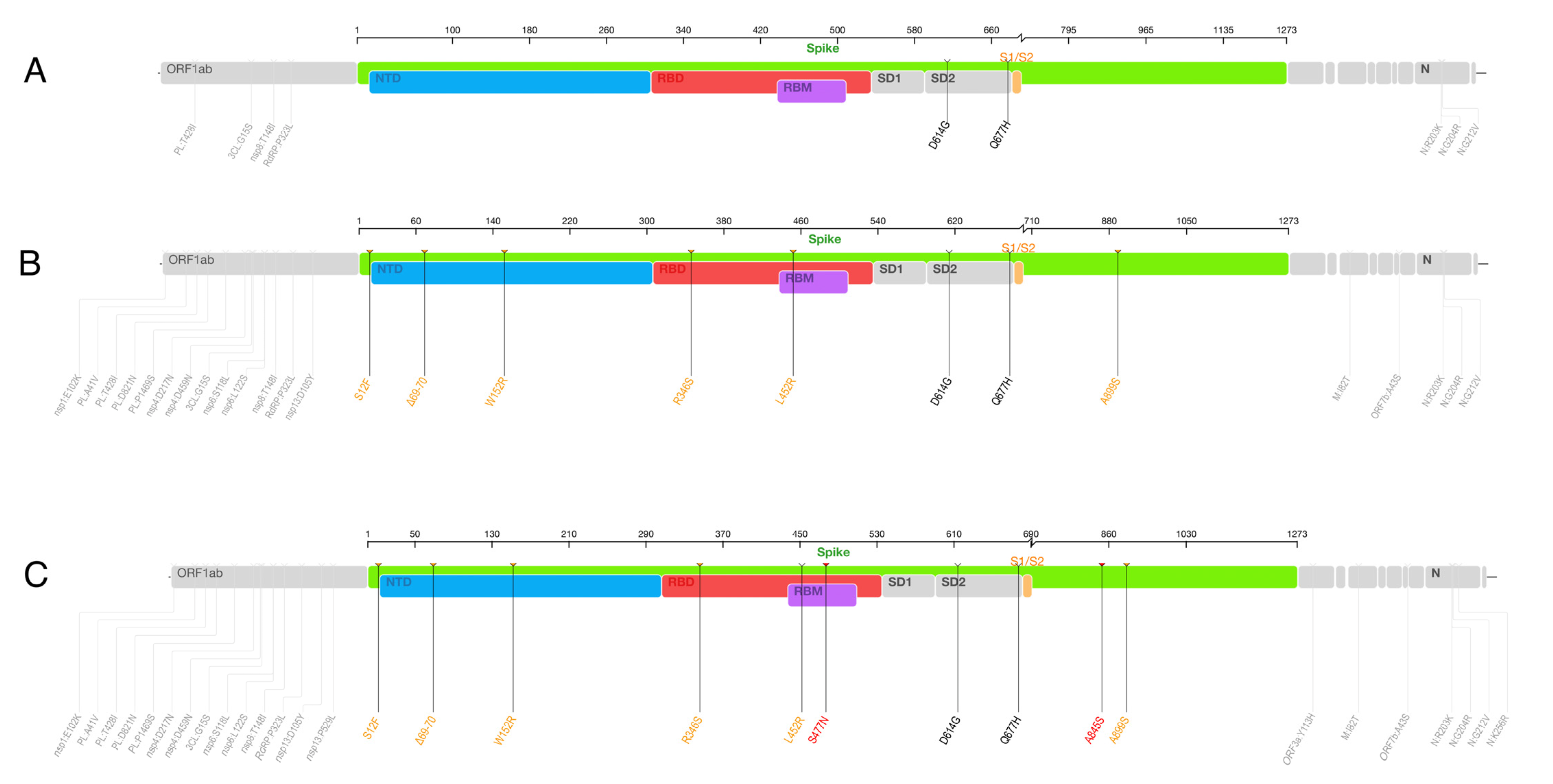
3.5. Mutational and Selection Analysis of the C.36.3 Lineage

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- ECDC. SARS-CoV-2 Variants of Concern as of 18 November 2021. Available online: https://www.ecdc.europa.eu/en (accessed on 9 August 2021).
- Gomaa, M.R.; El Rifay, A.S.; Shehata, M.; Kandeil, A.; Nabil Kamel, M.; Marouf, M.A.; GabAllah, M.; El Taweel, A.; Kayed, A.E.; Kutkat, O.; et al. Incidence, household transmission, and neutralizing antibody seroprevalence of Coronavirus Disease 2019 in Egypt: Results of a community-based cohort. PLoS Pathog. 2021, 17, e1009413. [Google Scholar] [CrossRef] [PubMed]
- Kandeil, A.; Mostafa, A.; El-Shesheny, R.; Shehata, M.; Roshdy, W.H.; Ahmed, S.S.; Gomaa, M.; Taweel, A.E.; Kayed, A.E.; Mahmoud, S.H.; et al. Coding-Complete Genome Sequences of Two SARS-CoV-2 Isolates from Egypt. Microbiol. Resour. Announc. 2020, 9, e00489-20. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. COVID-19 Weekly Epidemiological Update 2021; World Health Organization: Geneva, Switzerland, 2021.
- Our World in Data, O. COVID-19 Dataset by Our World in Data. Available online: https://github.com/owid/covid-19-data (accessed on 29 September 2021).
- Huisman, J.S.; Scire, J.; Angst, D.C.; Li, J.; Neher, R.A.; Maathuis, M.H.; Bonhoeffer, S.; Stadler, T. Estimation and worldwide monitoring of the effective reproductive number of SARS-CoV-2. medRxiv 2021. [Google Scholar] [CrossRef]
- Huisman, J.S. Daily SARS-CoV-2 Re Values for Select Countries. Available online: https://github.com/covid-19-Re/dailyRe-Data (accessed on 6 October 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A.; Kelso, J. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Neher, R. Nextalign: Viral Genome Reference Alignment. Available online: https://github.com/neherlab/nextalign (accessed on 16 December 2021).
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. Corrigendum to: IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 2461. [Google Scholar] [CrossRef] [PubMed]
- Tavare, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Some Math. Quest. Biol./DNA Seq. Anal. 1986, 17, 57–86. [Google Scholar]
- Lemoine, F.; Domelevo Entfellner, J.B.; Wilkinson, E.; Correia, D.; Davila Felipe, M.; De Oliveira, T.; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessells, R.; Cuadros, D.; Martin, D.P.; Rasmussen, D.A.; Zekri, A.-R.N.; Sangare, A.K.; et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 2021, 374, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200. [Google Scholar] [CrossRef] [PubMed]
- Saied, A.A.; Metwally, A.A.; Madkhali, N.A.B.; Haque, S.; Dhama, K. Egypt’s COVID-19 Recent Happenings and Perspectives: A Mini-Review. Front. Public Health 2021, 9, 696082. [Google Scholar] [CrossRef] [PubMed]
- Girgis, S.A.; Hafez, H.M.; Elarab, H.E.; Sherif, B.; Sabry, M.H.; Afifi, I.; Hassan, F.E.; Reda, A.; Elsayed, S.; Mahmoud, A.; et al. SARS-CoV-2 PCR positivity rate and seroprevalence of related antibodies among a sample of patients in Cairo: Pre-wave 2 results of a screening program in a university hospital. PLoS ONE 2021, 16, e0254581. [Google Scholar] [CrossRef] [PubMed]
- El-Sokkary, R.H.; Daef, E.; El-Korashi, L.A.; Khedr, E.M.; Gad, D.; Mohamed-Hussein, A.; Zayed, N.E.; Mostafa, E.F.; Bahgat, S.M.; Hassany, S.M.; et al. Sero-prevalence of anti-SARS-CoV-2 antibodies among healthcare workers: A multicenter study from Egypt. J. Infect. Public Health 2021, 14, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Landier, J.; Paireau, J.; Rebaudet, S.; Legendre, E.; Lehot, L.; Fontanet, A.; Cauchemez, S.; Gaudart, J. Cold and dry winter conditions are associated with greater SARS-CoV-2 transmission at regional level in western countries during the first epidemic wave. Sci. Rep. 2021, 11, 12756. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Domman, D.B.; Snyder, D.J.; Oguntuyo, K.Y.; Van Diest, M.; Densmore, K.H.; Schwalm, K.C.; Femling, J.; Carroll, J.L.; Scott, R.S.; et al. Emergence in late 2020 of multiple lineages of SARS-CoV-2 Spike protein variants affecting amino acid position 677. medRxiv 2021. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanat, F.; Thapa, M.; Lei, T.; Ahmed, S.M.S.; Adelsberg, D.C.; Carreño, J.M.; Strohmeier, S.; Schmitz, A.J.; Zafar, S.; Zhou, J.Q.; et al. SARS-CoV-2 mRNA vaccination induces functionally diverse antibodies to NTD, RBD, and S2. Cell 2021, 184, 3936–3948.e3910. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Public Health England. SARS-CoV-2 Variants of Concern and Variants under Investigation in England; 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1009243/Technical_Briefing_20.pdf (accessed on 9 August 2021).
- Timmers, L.F.S.M.; Peixoto, J.V.; Ducati, R.G.; Bachega, J.F.R.; de Mattos Pereira, L.; Caceres, R.A.; Majolo, F.; da Silva, G.L.; Anton, D.B.; Dellagostin, O.A.; et al. SARS-CoV-2 mutations in Brazil: From genomics to putative clinical conditions. Sci. Rep. 2021, 11, 11998. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.-Y.; Zhang, Y.; Zhou, X.; Huang, K.; Qian, Y.; Leng, Y.; Yan, L.; Huang, B.; He, Y. Molecular Evolution of SARS-CoV-2 Structural Genes: Evidence of Positive Selection in Spike Glycoprotein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nelson-Sathi, S.; Umasankar, P.K.; Sreekumar, E.; Radhakrishnan Nair, R.; Joseph, I.; Nori, S.R.C.; Philip, J.S.; Prasad, R.; Navyasree, K.V.; Ramesh, S.; et al. Mutational landscape and in silico structure models of SARS-CoV-2 Spike Receptor Binding Domain reveal key molecular determinants for virus-host interaction. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Kubik, S.; Arrigo, N.; Bonet, J.; Xu, Z. Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential. Viruses 2021, 13, 2114. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e1220. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- El-Saadani, S.; Saleh, M.; Ibrahim, S.A. Quantifying non-communicable diseases’ burden in Egypt using State-Space model. PLoS ONE 2021, 16, e0245642. [Google Scholar] [CrossRef]
- Weigang, S.; Fuchs, J.; Zimmer, G.; Schnepf, D.; Kern, L.; Beer, J.; Luxenburger, H.; Ankerhold, J.; Falcone, V.; Kemming, J.; et al. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat. Commun. 2021, 12, 6405. [Google Scholar] [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e5178. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Lessells, R.J.; Giandhari, J.; Wolter, N.; Everatt, J.; Rambaut, A.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. medRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roshdy, W.H.; Khalifa, M.K.; San, J.E.; Tegally, H.; Wilkinson, E.; Showky, S.; Martin, D.P.; Moir, M.; Naguib, A.; Elguindy, N.; et al. SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic. Viruses 2022, 14, 1878. https://0-doi-org.brum.beds.ac.uk/10.3390/v14091878
Roshdy WH, Khalifa MK, San JE, Tegally H, Wilkinson E, Showky S, Martin DP, Moir M, Naguib A, Elguindy N, et al. SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic. Viruses. 2022; 14(9):1878. https://0-doi-org.brum.beds.ac.uk/10.3390/v14091878
Chicago/Turabian StyleRoshdy, Wael H., Mohamed K. Khalifa, James Emmanuel San, Houriiyah Tegally, Eduan Wilkinson, Shymaa Showky, Darren Patrick Martin, Monika Moir, Amel Naguib, Nancy Elguindy, and et al. 2022. "SARS-CoV-2 Genetic Diversity and Lineage Dynamics in Egypt during the First 18 Months of the Pandemic" Viruses 14, no. 9: 1878. https://0-doi-org.brum.beds.ac.uk/10.3390/v14091878