
In Depth Viral Diversity Analysis in Atypical Neurological and Neonatal Chikungunya Infections in Rio de Janeiro, Brazil
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Samples
2.3. Viral RNA Isolation and Quantification
2.4. Virus Genome Deep Sequencing
2.5. Bioinformatics Data Processing
2.6. Phylogenetic Analysis
2.7. Viral Intrahost Genetic Diversity
2.8. Analyses in Silico
2.9. Statistical Analysis
3. Results and Discussion
3.1. Cases Selection and Characteristics
3.1.1. Vertical Transmission
Case 1
Case 2
Case 3
3.1.2. Neurological Manifestations
Case 1
Case 2
Case 3
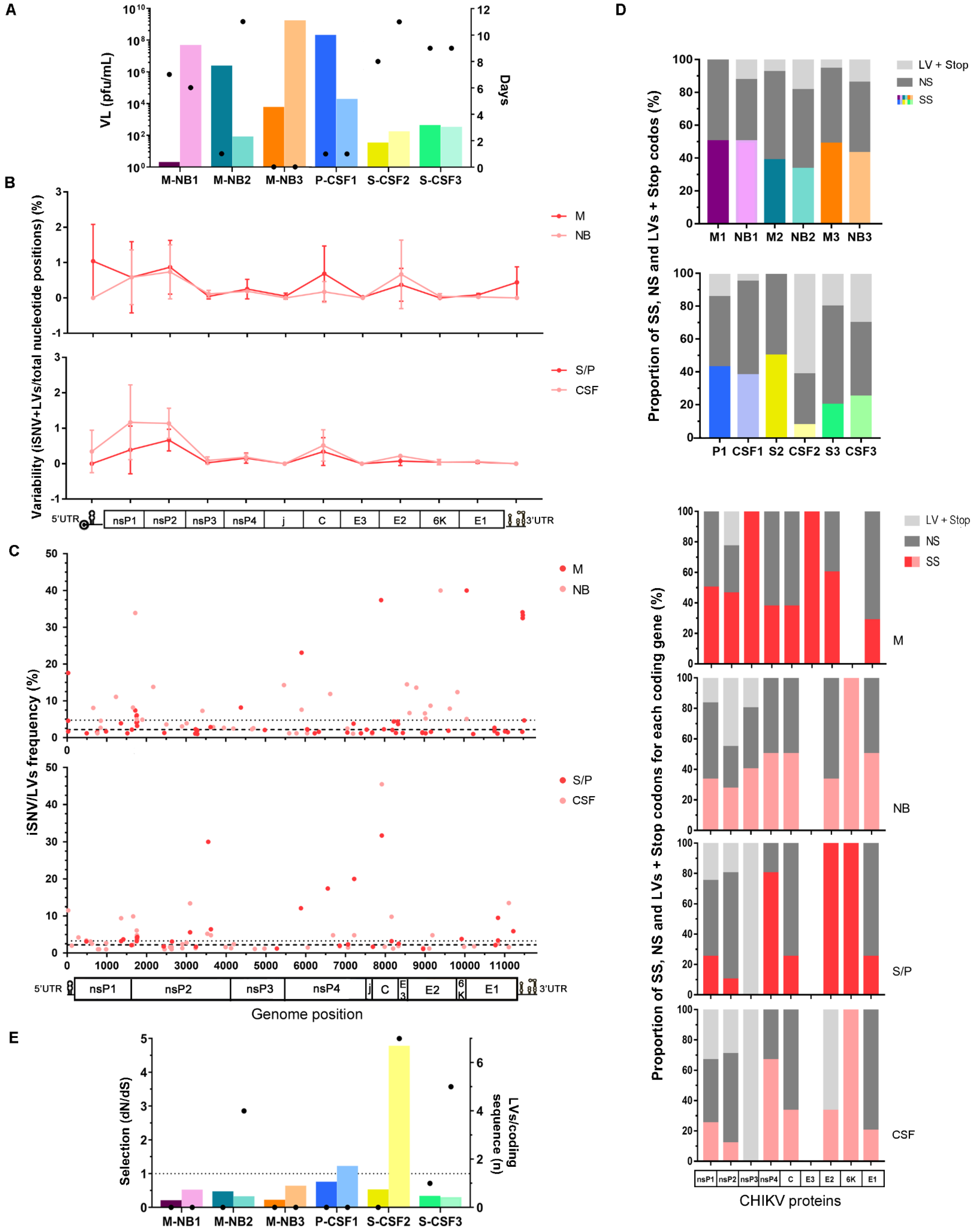
3.2. Intrahost CHIKV Genetic Diversity
3.3. Mutational Pattern
3.4. In Silico Analysis of Missense Mutations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Griffin, D.E. Alphaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2015; pp. 651–686. [Google Scholar]
- Yactayo, S.; Staples, J.E.; Millot, V.; Cibrelus, L.; Ramon-Pardo, P. Epidemiology of Chikungunya in the Americas. J. Infect Dis. 2016, 214 (Suppl. 5), S441–S445. [Google Scholar] [CrossRef] [PubMed]
- Bonifay, T.; Prince, C.; Neyra, C.; Demar, M.; Rousset, D.; Kallel, H.; Nacher, M.; Djossou, F.; Etpelboin, L. Atypical and severe manifestations of chikungunya virus infection in French Guiana: A hospital-based study. PLoS ONE 2018, 13, e0207406. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, T.J.; Guimarães, L.F.; Silva, M.T.; Soares, C.N. Neurological manifestations of Chikungunya and Zika infections. Arq. Neuropsiquiatr. 2016, 74, 937–943. [Google Scholar] [CrossRef]
- Mehta, R.; Gerardin, P.; de Brito, C.A.A.; Soares, C.N.; Ferreira, M.L.B.; Solomon, T. The neurological complications of chikungunya virus: A systematic review. Rev. Med. Virol. 2018, 28, e1978. [Google Scholar] [CrossRef]
- Das, T.; Jaffar-Bandjee, M.C.; Hoarau, J.J.; Krejbich Trotot, P.; Denizot, M.; Lee-Pat-Yuen, G.; Sahoo, R.; Guiraud, P.; Ramful, D.; Robin, S. Chikungunya fever: CNS infection and pathologies of a re-emerging arbovirus. Prog. Neurobiol. 2010, 91, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Arpino, C.; Curatolo, P.; Rezza, G. Chikungunya and the nervous system: What we do and do not know. Rev. Med. Virol. 2009, 19, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.S.; Costa, P.A.G.; Correa, I.A.; de Souza, M.R.M.; Calil, P.T.; da Silva, G.P.D.; Costa, S.M.; Fonseca, V.W.P.; da Costa, L.J. Chikungunya Virus: An Emergent Arbovirus to the South American Continent and a Continuous Threat to the World. Front. Microbiol. 2020, 11, 1297. [Google Scholar] [CrossRef] [PubMed]
- Traverse, E.M.; Millsapps, E.M.; Underwood, E.C.; Hopkins, H.K.; Young, M.; Barr, K.L. Chikungunya Immunopathology as It Presents in Different Organ Systems. Viruses 2022, 14, 1786. [Google Scholar] [CrossRef] [PubMed]
- Gerardin, P.; Barau, G.; Michault, A.; Bintner, M.; Randrianaivo, H.; Choker, G.; Lenglet, Y.; Touret, Y.; Bouveret, A.; Grivard, P.; et al. Multidisciplinary prospective study of mother-to-child chikungunya virus infections on the island of La Reunion. PLoS Med. 2008, 5, e60. [Google Scholar] [CrossRef]
- Gerardin, P.; Samperiz, S.; Ramful, D.; Boumahni, B.; Bintner, M.; Alessandri, J.-L.; Carbonnier, M.; Tiran-Rajaoefera, I.; Beullier, G.; Boya, I.; et al. Neurocognitive outcome of children exposed to perinatal mother-to-child chikungunya virus infection: The CHIMERE cohort study on Reunion Island. PLoS Negl. Trop. Dis. 2014, 8, e2996. [Google Scholar] [CrossRef]
- Fajardo, T.C.G.; Gazeta, R.E.; Catalan, D.T.; Mello, A.S.; Silva, A.C.B.D.; Bertozzi, A.P.A.P.; Dos Santos, G.R.; Pinto, C.A.L.; Monteiro, C.O.; Machado, R.R.G.; et al. Neonatal consequences of ma-ternal exposure to the chikungunya virus: Case reports. Medicine 2021, 100, e25695. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, O.C.C. The placenta in a case of pregnant woman infected by chikungunya virus. J. Virol. Retrovirol. 2016, 2, 1–4. [Google Scholar]
- Holmes, E. The RNA Virus Quasispecies. In The Evolution and Emergence of RNA Viruses; Oxford Series in Ecology and Evolution; Harvey, P.H., May, R.M., Eds.; Oxford University Press: Oxford, UK, 2009; pp. 87–103. [Google Scholar]
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef]
- Lee, H.Y.; Perelson, A.S.; Park, S.C.; Leitner, T. Dynamic correlation between intrahost HIV-1 quasispecies evolution and disease progression. PLoS Comput. Biol. 2008, 4, e1000240. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.G.; Bruden, D.; Deubner, H.; McArdle, S.; Chung, M.; Christensen, C.; Hennessy, T.; Homan, C.; Williams, J.; McMahon, B.J.; et al. Hepatitis C virus dynamics during natural infection are associated with long-term histological outcome of chronic hepatitis C disease. J. Infect Dis. 2007, 196, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef]
- Torres, M.C.; de Mendonça, M.C.L.; Rodrigues, C.D.D.S.; Fonseca, V.; Ribeiro, M.S.; Brandão, A.P.; da Cunha, R.V.; Dias, A.; Boas, L.S.V.; Felix, A.; et al. Dengue virus serotype 2 intrahost diversity in patients with different clinical outcomes. Viruses 2021, 13, 349. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Panella, A.J.; Velez, J.O.; Lambert, A.J.; Campbell, G.L. Chikungunya virus in US travelers re-turning from India, 2006. Emerg. Infect Dis. 2007, 13, 764–767. [Google Scholar] [CrossRef]
- Matranga, C.B.; Gladden-Young, A.; Qu, J.; Winnicki, S.; Nosamiefan, D.; Levin, J.Z.; Sabeti, P.C. Unbiased Deep Sequencing of RNA Viruses from Clinical Samples. J. Vis. Exp. 2016, 113, e54117. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Fauver, J.R.; Rückert, C.; Weger-Lucarelli, J.; Garcia-Luna, S.; Murrieta, R.A.; Gendernalik, A.; Smith, D.R.; Brackney, D.E.; Ebel, G.D. Mosquitoes Transmit Unique West Nile Virus Populations during Each Feeding Episode. Cell Rep. 2017, 19, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Del Barrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 1, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Fabri, A.A.; Rodrigues, C.D.D.S.; Santos, C.C.D.; Chalhoub, F.L.L.; Sampaio, S.A.; Faria, N.R.D.C.; Torres, M.C.; Fonseca, V.; Brasil, P.; Calvet, G.; et al. Co-Circulation of Two Independent Clades and Persistence of CHIKV-ECSA Genotype during Epidemic Waves in Rio de Janeiro, Southeast Brazil. Pathogens 2020, 9, 984. [Google Scholar] [CrossRef]
- Combe, M.; Garijo, R.; Geller, R.; Cuevas, J.M.; Sanjuán, R. Single-Cell Analysis of RNA Virus Infection Identifies Multiple Genetically Diverse Viral Genomes within Single Infectious Units. Cell Host Microbe 2015, 18, 424–432. [Google Scholar] [CrossRef]
- Vignuzzi, M.; López, C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- Salimi, H.; Cain, M.D.; Jiang, X.; Roth, R.A.; Beatty, W.L.; Sun, C.; Klimstra, W.B.; Hou, J.; Klein, R.S. Encephalitic alphaviruses exploit caveola-mediated transcytosis at the blood-brain barrier for central nervous system entry. MBio 2020, 11, e02731-19. [Google Scholar] [CrossRef]
- Ferguson, M.C.; Saul, S.; Fragkoudis, R.; Weisheit, S.; Cox, J.; Patabendige, A.; Sherwood, K.; Watson, M.; Merits, A.; Fazakerley, J.K. Ability of the encephalitic arbovirus Semliki Forest virus to cross the blood brain barrier is determined by the charge of the E2 glycoprotein. J. Virol. 2015, 89, 7536–7549. [Google Scholar] [CrossRef]
- Couderc, T.; Lecuit, M. Focus on Chikungunya pathophysiology in human and animal models. Microbes-Fection 2009, 11, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Grivard, P.; Le Roux, K.; Laurent, P.; Fianu, A.; Perrau, J.; Gigan, J.; Hoarau, G.; Grondin, N.; Staikowsky, F.; Favier, F.; et al. Molecular and serological diagnosis of Chikungunya virus infection. Pathol. Biol. 2007, 55, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Prata-Barbosa, A.; Cleto-Yamane, T.L.; Robaina, J.R.; Guastavino, A.B.; de Magalhães-Barbosa, M.C.; Brindeiro, R.D.M.; Medronhob, R.A.; Cunhaab, A.J.L.A. Co-infection with Zika and Chikungunya viruses associated with fetal death—A case report. Int. J. Infect Dis. 2018, 72, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Weger-Lucarelli, J.; Murrieta, R.A.; Fauver, J.R.; Garcia-Luna, S.M.; Prasad, A.N.; Black, W.C.; Ebel, G.D. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host Switching. Cell Host Microbe 2016, 19, 481–492. [Google Scholar] [CrossRef]
- Kollmann, T.R.; Kampmann, B.; Mazmanian, S.K.; Marchant, A.; Levy, O. Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 2017, 46, 350–363. [Google Scholar] [CrossRef]
- Owens, G.P. A Neuroprimer: Principles of Central Nervous System Immunity. Semin. Pediatr. Neurol. 2017, 24, 145–151. [Google Scholar] [CrossRef]
- Xiao, Y.; Dolan, P.T.; Goldstein, E.F.; Li, M.; Farkov, M.; Brodsky, L.; Andino, R. Poliovirus intrahost evolution is required to overcome tissue-specific innate immune responses. Nat. Commun. 2017, 8, 375. [Google Scholar] [CrossRef]
- Jones, R.; Bragagnolo, G.; Arranz, R.; Reguera, J. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 2021, 589, 615–619. [Google Scholar] [CrossRef]
- Singh, A.; Kumar, A.; Yadav, R.; Uversky, V.N.; Giri, R. Deciphering the dark proteome of Chikungunya virus. Sci. Rep. 2018, 8, 5822. [Google Scholar] [CrossRef]
- Fros, J.J.; Liu, W.J.; Prow, N.A.; Geertsema, C.; Ligtenberg, M.; Vanlandingham, D.L.; Schnettler, E.; Vlak, J.M.; Suhrbier, A.; Khromykh, A.; et al. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J. Virol. 2010, 84, 10877–10887. [Google Scholar] [CrossRef]
- Voss, J.E.; Vaney, M.C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Narwal, M.; Singh, H.; Pratap, S.; Malik, A.; Kuhn, R.J.; Kumar, P.; Tomar, S. Crystal structure of chikungunya virus nsP2 cysteine protease reveals a putative flexible loop blocking its active site. Int. J. Biol. Macromol. 2018, 116, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Law, Y.S.; Utt, A.; Tan, Y.B.; Zheng, J.; Wang, S.; Chen, M.W.; Griffin, P.R.; Merits, A.; Luo, D. Structural insights into RNA recognition by the Chikungunya virus nsP2 helicase. Proc. Natl. Acad. Sci. USA 2019, 116, 9558–9567. [Google Scholar] [CrossRef] [PubMed]
- Basore, K.; Kim, A.S.; Nelson, C.A.; Zhang, R.; Smith, B.K.; Uranga, C.; Vang, L.; Cheng, M.; Gross, M.L.; Smith, J.; et al. Cryo-EM Structure of Chikungunya Virus in Complex with the Mxra8 Receptor. Cell 2019, 177, 1725–1737.e16. [Google Scholar] [CrossRef]
- Kendall, C.; Khalid, H.; Müller, M.; Banda, D.H.; Kohl, A.; Merits, A.; Stonehouse, N.J.; Tuplin, A. Structural and phenotypic analysis of Chikungunya virus RNA replication elements. Nucleic Acids Res. 2019, 47, 9296–9312. [Google Scholar] [CrossRef]
- Lello, L.S.; Utt, A.; Bartholomeeusen, K.; Wang, S.; Rausalu, K.; Kendall, C.; Coppens, S.; Fragkoudis, R.; Tuplin, A.; Alphey, L.; et al. Cross-utilisation of template RNAs by alphavirus replicases. PLoS Pathog. 2020, 16, e1008825. [Google Scholar] [CrossRef]
- Torres, M.C.; Karl, A.L.M.; da Silva, M.M.P.; Dardenne, L.E.; de Filippis, A.M.B. In Silico Analysis of Dengue Virus Serotype 2 Mutations Detected at the Intrahost Level in Patients with Different Clinical Outcomes. Mi-crobiol Spectr. 2021, 9, e0025621. [Google Scholar] [CrossRef]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Region | Nt Substitution | Aa Substitution | 1M | 1RN | 2M | 2RN | 3M | 3R | P1 | CSF1 | S2 | CSF2 | S3 | CSF3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nsP1 | C482T | H136Y | 3.1 | 3.4 | ||||||||||
| A1355C | K427Q | 3.9 | 3.2 | 9.4 | ||||||||||
| G1660T | Q528H | 8.2 | 9.9 | |||||||||||
| nsP2 | G1748A | E23K | 4.1 | 5.9 | 3.3 | 4.4 | 6.1 | 3.2 | ||||||
| T2171C | F164L | x | x | |||||||||||
| T2625C | L315P | 1.0 | 1.6 | |||||||||||
| C3090T | T470I | 5.6 | 13.4 | |||||||||||
| A3467G | N596D | x | x | |||||||||||
| A3525G | E615G | x | ||||||||||||
| G3614A | V645T | 2.9 | 4.8 | 6.4 | ||||||||||
| nsP3 | C5631T | T519M | x | x | ||||||||||
| nsP4 | C6857T | H398Y | x | x | ||||||||||
| C7218T | T518I | 3.8 | 20.0 | 4.8 | ||||||||||
| E2 | A9035T | E165V | 5.2 | 1.2 | ||||||||||
| E1 | T11104G | S371A | 1.5 | 1.6 | 13.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, M.C.; Di Maio, F.; Brown, D.; Spyer, M.; Nastouli, E.; Brasil, P.; Bispo de Filippis, A.M. In Depth Viral Diversity Analysis in Atypical Neurological and Neonatal Chikungunya Infections in Rio de Janeiro, Brazil. Viruses 2022, 14, 2006. https://0-doi-org.brum.beds.ac.uk/10.3390/v14092006
Torres MC, Di Maio F, Brown D, Spyer M, Nastouli E, Brasil P, Bispo de Filippis AM. In Depth Viral Diversity Analysis in Atypical Neurological and Neonatal Chikungunya Infections in Rio de Janeiro, Brazil. Viruses. 2022; 14(9):2006. https://0-doi-org.brum.beds.ac.uk/10.3390/v14092006
Chicago/Turabian StyleTorres, Maria Celeste, Fatima Di Maio, David Brown, Moira Spyer, Eleni Nastouli, Patrícia Brasil, and Ana Maria Bispo de Filippis. 2022. "In Depth Viral Diversity Analysis in Atypical Neurological and Neonatal Chikungunya Infections in Rio de Janeiro, Brazil" Viruses 14, no. 9: 2006. https://0-doi-org.brum.beds.ac.uk/10.3390/v14092006