
Inflammatory Control of Viral Infection

1
Medical Microbiology and Immunology, University of Toledo College of Medicine and Life Sciences, Toledo, OH 43614, USA
2
Physiology and Pharmacology, University of Toledo College of Medicine and Life Sciences, Toledo, OH 43614, USA
*
Author to whom correspondence should be addressed.
Viruses 2023, 15(7), 1579; https://0-doi-org.brum.beds.ac.uk/10.3390/v15071579
Submission received: 25 June 2023
/
Revised: 18 July 2023
/
Accepted: 19 July 2023
/
Published: 20 July 2023
(This article belongs to the Special Issue Interferons in Viral Infections)
{kind=link}
{kind=link}
Abstract
:Inflammatory responses during virus infection differentially impact the host. Managing inflammatory responses is essential in controlling viral infection and related diseases. Recently, we identified a cellular anti-inflammatory mechanism, RIKA (Repression of IRF3-mediated inhibition of NF-κB activity), which controls viral inflammation and pathogenesis. The RIKA function of IRF3 may be explored further in other inflammatory diseases beyond viral infection.
Viral infection rapidly turns on a plethora of genes, many of which are critical for successful clearance of the virus. Interferons (IFNs) and IFN-stimulated genes (ISGs) are the most prominent ones that inhibit a wide range of viruses [1,2]. Virus-infected cells, in addition to antiviral genes, induce inflammatory genes, which are critical for innate immune responses. Deficiency or imbalance in antiviral or inflammatory arms of immune responses causes undesired outcomes, i.e., viral diseases. Cellular regulators are in place to control both sides of innate immune responses; carefully managing both arms can help avoid these undesired outcomes.
Viral components are detected in infected cells by pattern recognition receptors (PRRs), e.g., TLRs, RLRs, cGAS, etc. [3,4]. Upon binding viral nucleic acids, PRRs trigger downstream signaling pathways to activate IRF3 and NF-κB, transcription factors that cooperatively act to induce type-I as well as type-III IFNs. PRR-activated IRF3 causes expression of antiviral genes, whereas NF-κB activation leads to pro-inflammatory gene expression [5,6,7]. In the early phase, inflammatory genes help protect against infection; however, in the later stage, they can cause cell and tissue damage by inducing cytokine storm, cell death, etc. [8,9]. Inflammation has beneficial effects on tissue homeostasis and resolution of infection; however, harmful effects are not completely clear. It is largely appreciated that viral load is the predominant factor contributing to viral diseases. However, it is becoming increasingly clear, particularly from studies on SARS-CoV-2 and influenza A virus (IAV) infection, that virus-induced lung inflammation contributes significantly to viral pathogenesis [10,11,12,13]. Neuroinflammation by herpesviruses and flaviviruses, as well as cardiac inflammation by reoviruses, contribute to lethal viral diseases [14,15,16,17,18,19,20]. Controlling virus-induced inflammation when it is harmful to the host can suppress viral pathogenesis.
IRF3 is antiviral against a wide range of viruses; IRF3 deficiency causes increased viral replication and susceptibility to viral infection [21,22]. IRF3 activation causes the transcriptional induction of IFNβ, a cytokine, which is secreted from infected cells and acts by autocrine and paracrine signaling to trigger a transcriptional induction of ISGs [23,24]. For transcriptional activity, IRF3 is phosphorylated by TBK1, allowing its translocation into the nucleus. In addition to the transcriptional response, virus-activated IRF3 causes apoptotic cell death in a transcription-independent pathway, RLR-induced IRF3-mediated Pathway of Apoptosis (RIPA, Figure 1) [25,26,27,28]. For RIPA, IRF3 undergoes linear ubiquitination by LUBAC, leading to its interaction with pro-apoptotic protein BAX, causing translocation into mitochondria. An IRF3 mutant, defective in transcriptional response but active in RIPA, can inhibit viral replication in cells and mice.
Recently, we uncovered a new function of IRF3, Repression of IRF3-mediated NF-κB Activation (RIKA), inhibiting NF-κB activity to suppress inflammatory gene expression (Figure 1) [29]. IRF3 knockout cells express elevated NF-κB-dependent genes, e.g., the inflammatory target genes, upon Sendai virus (SeV), influenza A virus (IAV), and murine hepatitis virus (MHV) infection compared to the control cells. Microarray analyses using control and IRF3-knockdown cells further validated these results. In addition to virus infection, we used non-viral stimuli, e.g., polyI:C, LPS, and cGAMP, to demonstrate a RIKA-mediated suppression of inflammatory genes. Mechanistically, IRF3 interacts with the NF-κB-p65 subunit directly, sequestering in cytosol to inhibit its translocation into the nucleus. Using deletion analyses, we mapped the domains responsible for the IRF3:NF-κB-p65 interaction. IRF3 mutants, defective in either transcriptional activity (IRF3-S1), or RIPA, or both (IRF3-M1), were able to interact with NF-κB-p65 and exhibit RIKA (Figure 2). Surprisingly, IRF3-M1, like IRF3-Wt, inhibits viral replication, demonstrating the antiviral activity of RIKA. Finally, IRF3 knockout mice, infected intranasally with SeV, exhibit enhanced inflammatory genes in the infected lungs compared to Wt mice. Therefore, virus-induced inflammation is a critical determinant of viral pathogenesis, and the RIKA branch of IRF3 contributes to the optimal antiviral activity of IRF3.
Although discovered in the context of virus infection, RIKA has implications beyond viral diseases. Many bacteria rely on cellular inflammatory responses, and RIKA may promote their replication and pathogenesis. RIKA plays a key role in alleviating high-fat diet (HFD)-induced liver injury [30]. In the HFD-induced liver injury model, IRF3 interacts with NF-κB-p65, inhibiting its activation and inflammatory gene expression. Knock-in mice, expressing the IRF3-S1 mutant, transcriptionally inactive but active in RIKA, suppress HFD-induced liver injury. A RIKA-like activity of IRF3 controls HFD-induced hepatic steatosis and insulin resistance, in which IRF3 interacts with IKKβ to suppress hepatic inflammatory responses [31]. IRF3 promotes LPS-induced septic shock in mice; IRF3 knockout mice are resistant to LPS-mediated sepsis compared to Wt mice [32,33]. However, the molecular mechanisms are unclear; whether RIKA-like activities play any role in the sepsis-promoting function of IRF3 needs to be investigated. Many cancer cells rely on NF-κB activity for survival since NF-κB is involved in anti-apoptotic gene expression [34,35,36]. The RIKA function of IRF3 may be explored for inhibiting NF-κB activity to promote cancer cell death. Future studies involving pathway-specific IRF3 mutants (as shown in Figure 2) can be utilized to elucidate the role of RIKA in diseases beyond viral infection. IRF3-derived small peptides, designed to specifically bind NF-κB, may have anti-inflammatory activities, with therapeutic potential against inflammatory diseases.
A major question that remains to be answered is whether these pathways of IRF3 operate simultaneously or sequentially in the infected cells. It is speculated that in the early stage, cells would primarily rely on the transcriptional pathway of IRF3 to mount antiviral responses, which also require NF-κB activity. In the second phase, RIKA may be critical to curb the undesired inflammatory responses. Later in the infection, as we have also shown in SeV-infected cells, RIPA-mediated apoptotic killing can help eliminate the infected cells. It remains to be seen whether these pathways are operational in all cells or if there is cell type-specific activation of IRF3 functions. The pathway-specific mutants will be critical tools to tease them apart in future studies. Finally, our investigation can be expanded to other IRFs and viral IRFs to examine the specificity of IRF-dependent cellular anti-inflammatory responses. In summary, the identification of RIKA opens new avenues for addressing various critical questions related to IRF biology.
Author Contributions
Conceptualization, S.C. (Saurabh Chattopadhyay) and S.C. (Sukanya Chakravarty); writing, S.C. (Sukanya Chakravarty), S.C. (Saurabh Chattopadhyay) and R.C. All authors have read and agreed to the published version of the manuscript.
Funding
Research in authors’ laboratory is funded by National Institutes of Health grants AI155545, AI165521.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Crosse, K.M.; Monson, E.A.; Beard, M.R.; Helbig, K.J. Interferon-Stimulated Genes as Enhancers of Antiviral Innate Immune Signaling. J. Innate Immun. 2018, 10, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Lukhele, S.; Boukhaled, G.M.; Brooks, D.G. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol. 2019, 43, 101277. [Google Scholar] [CrossRef]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e117. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. Innate immunity, cytokine storm, and inflammatory cell death in COVID-19. J. Transl. Med. 2022, 20, 542. [Google Scholar] [CrossRef]
- Gu, Y.; Zuo, X.; Zhang, S.; Ouyang, Z.; Jiang, S.; Wang, F.; Wang, G. The Mechanism behind Influenza Virus Cytokine Storm. Viruses 2021, 13, 1362. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.H.; Yang, Z.Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm—The common denominator and the lessons to be learned. Clin. Immunol. 2021, 223, 108652. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zheng, K. Microglial mitophagy integrates the microbiota-gut-brain axis to restrain neuroinflammation during neurotropic herpesvirus infection. Autophagy 2023, 19, 734–736. [Google Scholar] [CrossRef]
- Leibovitch, E.C.; Caruso, B.; Ha, S.K.; Schindler, M.K.; Lee, N.J.; Luciano, N.J.; Billioux, B.J.; Guy, J.R.; Yen, C.; Sati, P.; et al. Herpesvirus trigger accelerates neuroinflammation in a nonhuman primate model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 11292–11297. [Google Scholar] [CrossRef] [Green Version]
- Wainberg, M.; Luquez, T.; Koelle, D.M.; Readhead, B.; Johnston, C.; Darvas, M.; Funk, C.C. The viral hypothesis: How herpesviruses may contribute to Alzheimer’s disease. Mol. Psychiatry 2021, 26, 5476–5480. [Google Scholar] [CrossRef]
- Pan, Y.; Cai, W.; Cheng, A.; Wang, M.; Yin, Z.; Jia, R. Flaviviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Front. Immunol. 2022, 13, 829433. [Google Scholar] [CrossRef] [PubMed]
- van Marle, G.; Antony, J.; Ostermann, H.; Dunham, C.; Hunt, T.; Halliday, W.; Maingat, F.; Urbanowski, M.D.; Hobman, T.; Peeling, J.; et al. West Nile virus-induced neuroinflammation: Glial infection and capsid protein-mediated neurovirulence. J. Virol. 2007, 81, 10933–10949. [Google Scholar] [CrossRef] [Green Version]
- Mantri, M.; Hinchman, M.M.; McKellar, D.W.; Wang, M.F.Z.; Cross, S.T.; Parker, J.S.; De Vlaminck, I. Spatiotemporal transcriptomics reveals pathogenesis of viral myocarditis. Nat. Cardiovasc. Res. 2022, 1, 946–960. [Google Scholar] [CrossRef]
- Dina Zita, M.; Phillips, M.B.; Stuart, J.D.; Kumarapeli, A.R.; Snyder, A.J.; Paredes, A.; Sridharan, V.; Boerma, M.; Danthi, P.; Boehme, K.W. The M2 Gene Is a Determinant of Reovirus-Induced Myocarditis. J. Virol. 2022, 96, e0187921. [Google Scholar] [CrossRef]
- Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Glanz, A.; Chakravarty, S.; Varghese, M.; Kottapalli, A.; Fan, S.; Chakravarti, R.; Chattopadhyay, S. Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus. Viruses 2021, 13, 575. [Google Scholar] [CrossRef]
- Ikushima, H.; Negishi, H.; Taniguchi, T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold. Spring Harb. Symp. Quant. Biol. 2013, 78, 105–116, Research Support, Non-U.S. Gov’t. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Kuzmanovic, T.; Zhang, Y.; Wetzel, J.L.; Sen, G.C. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 2016, 44, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Glanz, A.; Chakravarty, S.; Fan, S.; Chawla, K.; Subramanian, G.; Rahman, T.; Walters, D.; Chakravarti, R.; Chattopadhyay, S. Autophagic degradation of IRF3 induced by the small-molecule auranofin inhibits its transcriptional and proapoptotic activities. J. Biol. Chem. 2021, 297, 101274. [Google Scholar] [CrossRef] [PubMed]
- Glanz, A.; Chawla, K.; Fabry, S.; Subramanian, G.; Garcia, J.; Jay, B.; Ciricillo, J.; Chakravarti, R.; Taylor, R.T.; Chattopadhyay, S. High Throughput Screening of FDA-Approved Drug Library Reveals the Compounds that Promote IRF3-Mediated Pro-Apoptotic Pathway Inhibit Virus Replication. Viruses 2020, 12, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popli, S.; Chakravarty, S.; Fan, S.; Glanz, A.; Aras, S.; Nagy, L.E.; Sen, G.C.; Chakravarti, R.; Chattopadhyay, S. IRF3 inhibits nuclear translocation of NF-kappaB to prevent viral inflammation. Proc. Natl. Acad. Sci. USA 2022, 119, e2121385119. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, C.; McMullen, M.R.; Chattopadhyay, S.; Roychowdhury, S.; Sen, G.; Nagy, L.E. Nontranscriptional Activity of Interferon Regulatory Factor 3 Protects Mice From High-Fat Diet-Induced Liver Injury. Hepatol. Commun. 2019, 3, 1626–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.A.; Zhang, R.; She, Z.G.; Zhang, X.F.; Jiang, D.S.; Wang, T.; Gao, L.; Deng, W.; Zhang, S.M.; Zhu, L.H.; et al. Interferon regulatory factor 3 constrains IKKbeta/NF-kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 2014, 59, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Chiba, S.; Hangai, S.; Kometani, K.; Inoue, A.; Kimura, Y.; Abe, T.; Kiyonari, H.; Nishio, J.; Taguchi-Atarashi, N.; et al. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 5253–5258. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Fensterl, V.; Zhang, Y.; Veleeparambil, M.; Wetzel, J.L.; Sen, G.C. Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF-3 are regulated by the acetylation and phosphorylation of its coactivators. MBio 2013, 4, e00636, Research Support, N.I.H., Extramural. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, M.; Matos, P.; Pereira, T.; Cabrera, R.; Cardoso, B.A.; Bugalho, M.J.; Silva, A.L. RAC1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS ONE 2017, 12, e0172689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacifico, F.; Leonardi, A. Role of NF-kappaB in thyroid cancer. Mol. Cell. Endocrinol. 2010, 321, 29–35. [Google Scholar] [CrossRef]
- Man, X.; Piao, C.; Lin, X.; Kong, C.; Cui, X.; Jiang, Y. Correction to: USP13 functions as a tumor suppressor by blocking the NF-kB-mediated PTEN downregulation in human bladder cancer. J. Exp. Clin. Cancer Res. 2021, 40, 386. [Google Scholar] [CrossRef]
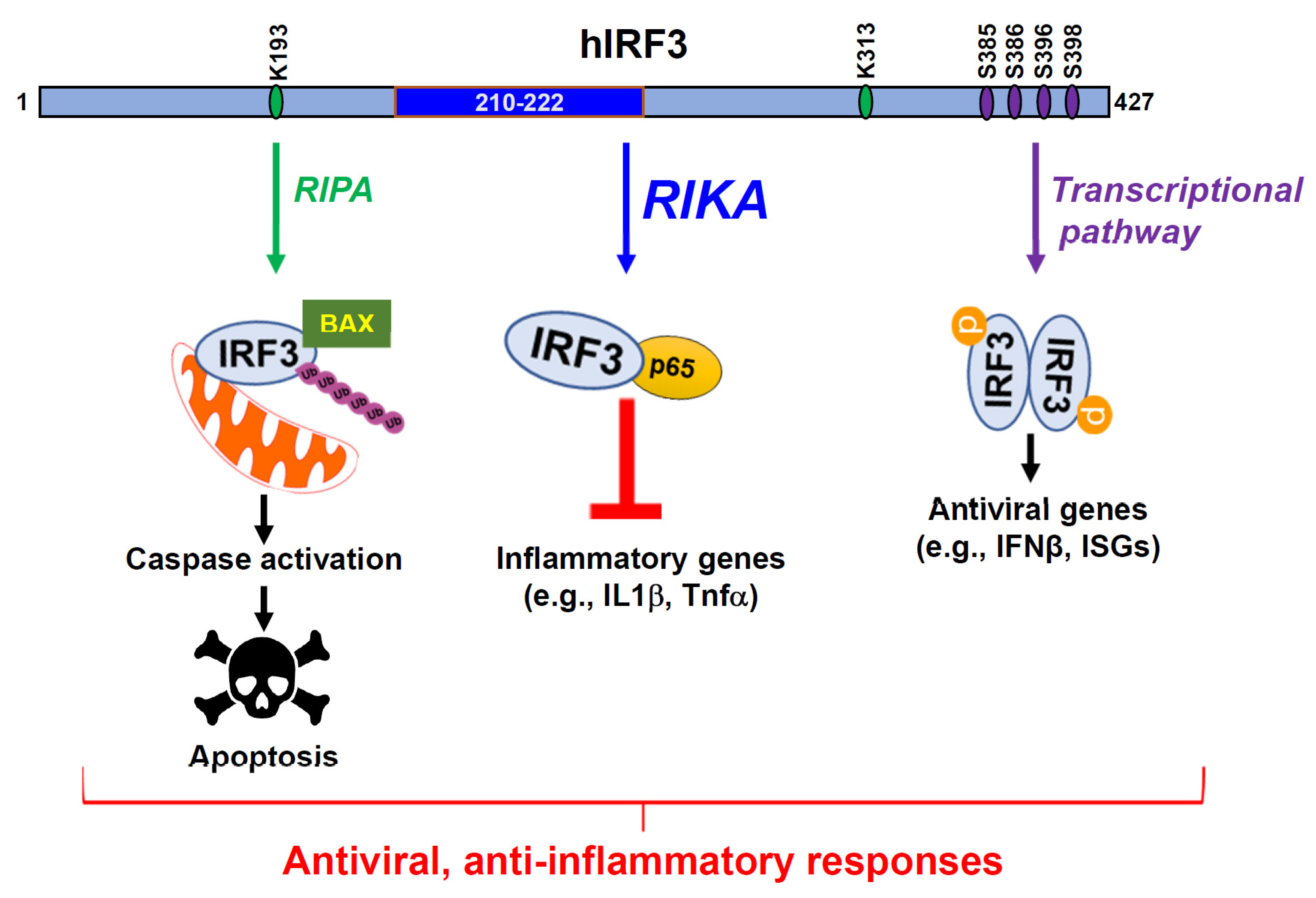
Figure 1.
RIKA, a new anti-inflammatory function of IRF3. IRF3 is widely known for its transcriptional function involving the production of IFNβ and ISGs which antagonize various stages of viral replication. A non-transcriptional function of IRF3, RIPA (RIG-I-like receptor-induced IRF3 mediated Pathway of Apoptosis), was uncovered through our studies, involving IRF3 apoptotically killing virus-infected cells. In RIPA, linear ubiquitination of IRF3 upon virus infection results in its activation and translocation into the mitochondria. Subsequent activation of downstream caspases results in apoptotic cell death. RIKA, a new non-transcriptional function of IRF3, downregulates inflammatory gene expression. IRF3 domain, comprising 210–222 amino acids, interacts with the p65 subunit of NF-κB to sequester it in the cytosol. As a result, induction of inflammatory cytokines such as IL1β, Tnfα is restricted, thus suppressing the inflammatory responses and pathogenesis caused by the overproduction of these cytokines.
Figure 1.
RIKA, a new anti-inflammatory function of IRF3. IRF3 is widely known for its transcriptional function involving the production of IFNβ and ISGs which antagonize various stages of viral replication. A non-transcriptional function of IRF3, RIPA (RIG-I-like receptor-induced IRF3 mediated Pathway of Apoptosis), was uncovered through our studies, involving IRF3 apoptotically killing virus-infected cells. In RIPA, linear ubiquitination of IRF3 upon virus infection results in its activation and translocation into the mitochondria. Subsequent activation of downstream caspases results in apoptotic cell death. RIKA, a new non-transcriptional function of IRF3, downregulates inflammatory gene expression. IRF3 domain, comprising 210–222 amino acids, interacts with the p65 subunit of NF-κB to sequester it in the cytosol. As a result, induction of inflammatory cytokines such as IL1β, Tnfα is restricted, thus suppressing the inflammatory responses and pathogenesis caused by the overproduction of these cytokines.

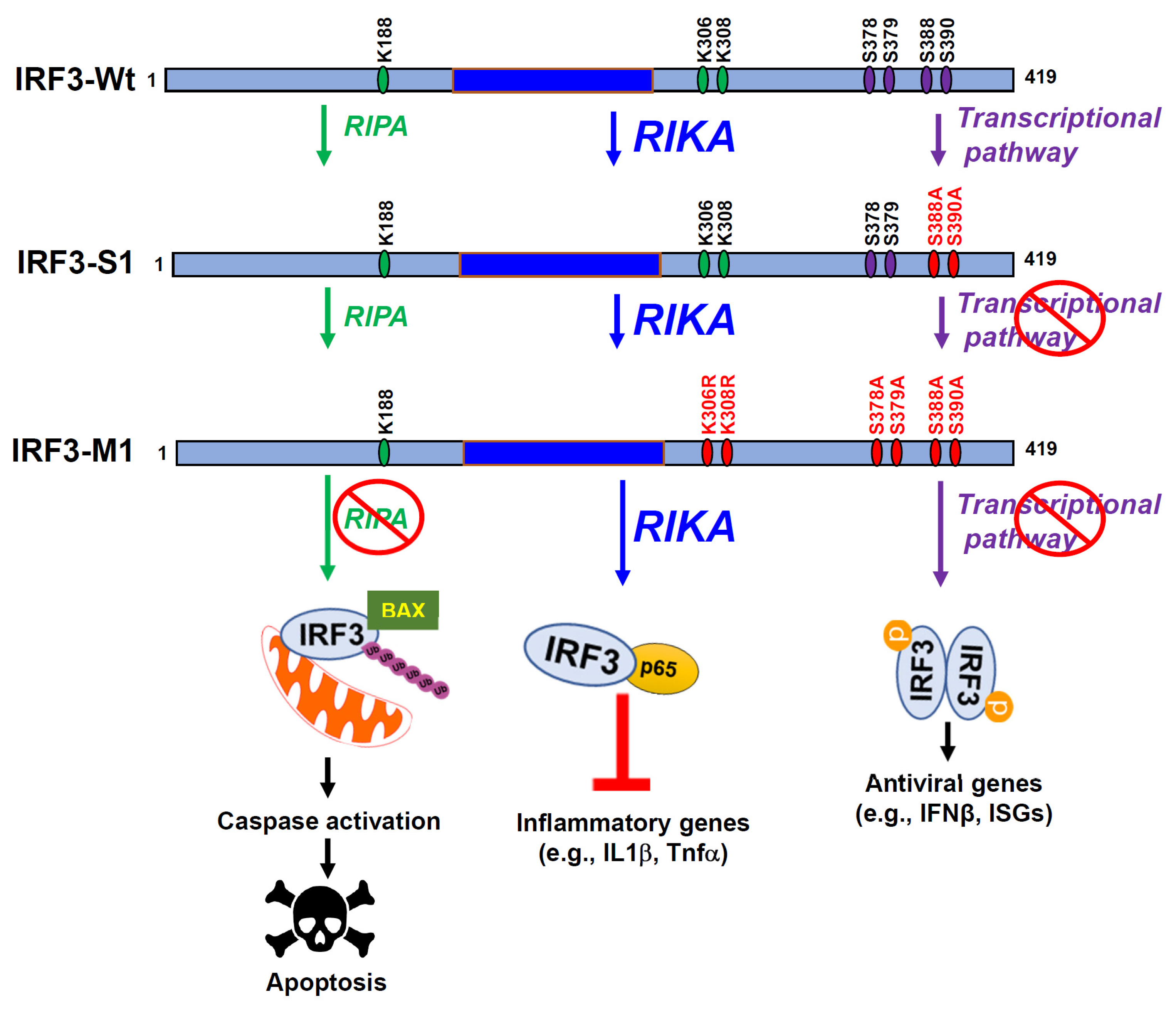
Figure 2.
Pathway-specific mutants of IRF3 and their functions. Mouse IRF3 (IRF3-Wt) with its critical amino acids and pathway-specific mutants (IRF3-S1 and IRF3-M1) are shown. Wt IRF3 is capable of transcriptional, RIPA, and RIKA functions, IRF3-S1 is active in RIPA and RIKA but inactive in transcriptional activity, and IRF3-M1 is active only in RIKA but inactive in transcriptional and RIPA activities.
Figure 2.
Pathway-specific mutants of IRF3 and their functions. Mouse IRF3 (IRF3-Wt) with its critical amino acids and pathway-specific mutants (IRF3-S1 and IRF3-M1) are shown. Wt IRF3 is capable of transcriptional, RIPA, and RIKA functions, IRF3-S1 is active in RIPA and RIKA but inactive in transcriptional activity, and IRF3-M1 is active only in RIKA but inactive in transcriptional and RIPA activities.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chakravarty, S.; Chakravarti, R.; Chattopadhyay, S. Inflammatory Control of Viral Infection. Viruses 2023, 15, 1579. https://0-doi-org.brum.beds.ac.uk/10.3390/v15071579
AMA Style
Chakravarty S, Chakravarti R, Chattopadhyay S. Inflammatory Control of Viral Infection. Viruses. 2023; 15(7):1579. https://0-doi-org.brum.beds.ac.uk/10.3390/v15071579
Chicago/Turabian StyleChakravarty, Sukanya, Ritu Chakravarti, and Saurabh Chattopadhyay. 2023. "Inflammatory Control of Viral Infection" Viruses 15, no. 7: 1579. https://0-doi-org.brum.beds.ac.uk/10.3390/v15071579
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.