

Deciphering the Role of Post-Translational Modifications and Cellular Location of Hepatitis Delta Virus (HDV) Antigens in HDV-Mediated Liver Damage in Mice
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site-Directed Mutagenesis (SDM)
2.2. Cell Lines
2.3. DNA Transfection
2.4. Generation of AAV Vectors
2.5. Animal Manipulation and Procedures
2.6. Protein Extraction from Cells and Liver Samples
2.7. Immunofluorescence (IF)
2.8. Determination of HDV Infectious Viral Particles
2.9. Western Blot
2.10. Histology and Immunohistochemistry (IHC)
2.11. RNA Extraction and RT-qPCR
2.12. Statistical Analysis ç
3. Results
3.1. Analysis of HDAgs Expression and HDV Replication in Huh-7 Cells Transfected with HDV WT and Mutants
3.2. Analysis of Intracellular Localization of HDAgs and Assessment of HDV Infectious Virion Formation
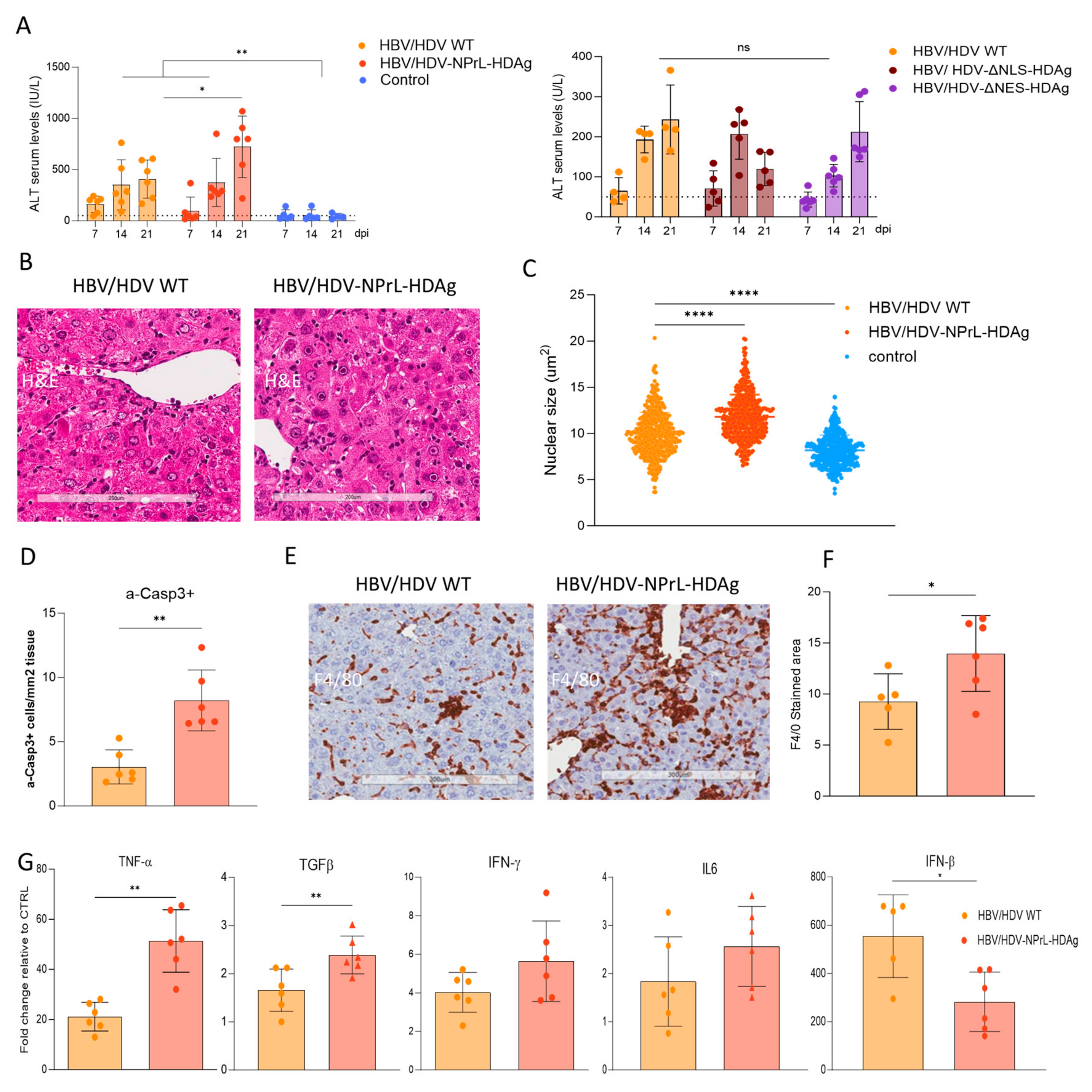
3.3. In Vivo Analysis of HDV-NPrL-HDAg, HDV-ΔNLS-HDAg, and HDV-ΔNES-HDAg Mutants
3.4. Evaluation of Liver Damage
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/Taxonomy/Browser/wwwtax.cgi (accessed on 19 February 2024).
- Bonino, F.; Heermann, K.H.; Rizzetto, M.; Gerlich, W.H. Hepatitis Delta Virus: Protein Composition of Delta Antigen and Its Hepatitis B Virus-Derived Envelope. J. Virol. 1986, 58, 945–950. [Google Scholar] [CrossRef]
- Kos, A.; Dijkema, R.; Arnberg, A.C.; van der Meide, P.H.; Schellekens, H. The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986, 323, 558–560. [Google Scholar] [CrossRef]
- Ryu, W.; Netter, H.J.; Bayer, M.; Taylor, J. Ribonucleoprotein Complexes of Hepatitis Delta Virus. J. Virol. 1993, 67, 3281–3287. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M. Chapter 3. Replication of the hepatitis delta virus RNA genome. Adv. Virus Res. 2009, 74, 103–121. [Google Scholar]
- Gudima, S.; Chang, J.; Moraleda, G.; Azvolinsky, A.; Taylor, J. Parameters of Human Hepatitis Delta Virus Genome Replication: The Quantity, Quality, and Intracellular Distribution of Viral Proteins and RNA. J. Virol. 2002, 76, 3709–3719. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M. Hepatitis Delta Virus: Cis and Trans Functions Required for Replication. Cell 1990, 61, 371–373. [Google Scholar] [CrossRef]
- Jeng, K.S.; Daniel, A.; Lai, M.M. A pseudoknot ribozyme structure is active in vivo and required for hepatitis delta virus RNA replication. J. Virol. 1996, 70, 2403–2410. [Google Scholar] [CrossRef]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef]
- Lee, C.Z.; Chen, P.J.; Chen, D.S. Large hepatitis delta antigen in packaging and replication inhibition: Role of the carboxyl-terminal 19 amino acids and amino-terminal sequences. J. Virol. 1995, 69, 5332–5336. [Google Scholar] [CrossRef]
- Chang, F.L.; Chen, P.J.; Tu, S.J.; Wang, C.J.; Chen, D.S. The large form of hepatitis delta antigen is crucial for assembly of hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1991, 88, 8490–8494. [Google Scholar] [CrossRef]
- Chang, M.; Chang, S.C.; Chang, C.; Wu, K. Nuclear Localization Signals, but Not Putative Leucine Zipper Motifs, Are Essential for Nuclear Transport of Hepatitis Delta Antigen. J. Virol. 1992, 66, 6019–6027. [Google Scholar] [CrossRef]
- Xia, Y.; Yeh, C.; Ou, J.; Lai, M.M.C. Characterization of Nuclear Targeting Signal of Hepatitis Delta Antigen: Nuclear Transport as a Protein Complex. J. Virol. 1992, 66, 914–921. [Google Scholar] [CrossRef]
- Alves, C.; Freitas, N.; Cunha, C. Characterization of the nuclear localization signal of the hepatitis delta virus antigen. Virology 2008, 370, 12–21. [Google Scholar] [CrossRef]
- Lee, C.H.; Chang, S.C.; Wu, C.H.H.; Chang, M.F. A Novel Chromosome Region Maintenance 1-independent Nuclear Export Signal of the Large Form of Hepatitis Delta Antigen That Is Required for the Viral Assembly. J. Biol. Chem. 2001, 276, 8142–8148. [Google Scholar] [CrossRef]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a Prenylation Site in Delta Virus Large Antigen. Science 1992, 256, 1331–1333. [Google Scholar] [CrossRef]
- Hwang, S.B.; Lai, M.M. Isoprenylation Mediates Direct Protein-Protein Interactions between Hepatitis Large Delta Antigen and Hepatitis B Virus Surface Antigen. J. Virol. 1993, 67, 7659–7662. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Chen, D.; Irol, J.V. The Conserved Serine 177 in the Delta Antigen of Hepatitis Delta Virus Is One Putative Phosphorylation Site and Is Required for Efficient Viral RNA Replication. J. Virol. 2001, 75, 9087–9095. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, W.; Hong, S.; Tsay, Y.; Chen, P. ERK1/2-Mediated Phosphorylation of Small Hepatitis Delta Antigen at Serine 177 Enhances Hepatitis Delta Virus Antigenomic RNA Replication. J. Virol. 2008, 82, 9345–9358. [Google Scholar] [CrossRef] [PubMed]
- Bichko, V.; Barik, S.; Taylor, J. Phosphorylation of the Hepatitis Delta Virus Antigens. J. Virol. 1997, 71, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.S.; Lee, Y.H.W. Assembly of hepatitis delta virus particles: Package of multimeric hepatitis delta virus genomic RNA and role of phosphorylation. Virology 1998, 249, 12–20. [Google Scholar] [CrossRef]
- Li, Y.; Macnaughton, T.; Gao, L.; Lai, M.C. RNA-Templated Replication of Hepatitis Delta Virus: Genomic and Antigenomic RNAs Associate with Different Nuclear Bodies. J. Virol. 2006, 80, 6478–6486. [Google Scholar] [CrossRef]
- Bichko, V.V.; Taylor, J.M. Redistribution of the Delta Antigens in Cells Replicating the Genome of Hepatitis Delta Virus. J. Virol. 1996, 70, 8064–8070. [Google Scholar] [CrossRef]
- Chang, M.F.; Baker, S.C.; Soe, L.H.; Kamahora, T.; Keck, J.G.; Makino, S.; Govindarajan, S.; Lai, M.M. Human Hepatitis Delta Antigen Is a Nuclear Phosphoprotein with RNA Binding Activity. J. Virol. 1998, 62, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Alves, C.; Gudima, S.; Taylor, J. Intracellular Localization of Hepatitis Delta Virus Proteins in the Presence and Absence of Viral RNA Accumulation. J. Virol. 2009, 83, 6457–6463. [Google Scholar] [CrossRef]
- Negro, F.; Lok, A.S. Hepatitis D: A Review. J. Am. Med. Assoc. 2023, 330, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Abdul Majeed, N.; Zehnder, B.; Koh, C.; Heller, T.; Urban, S. Hepatitis delta: Epidemiology to recent advances in therapeutic agents. Hepatology 2023, 78, 1306–1321. [Google Scholar] [CrossRef]
- Aldabe, R.; Suárez-Amarán, L.; Usai, C.; González-Aseguinolaza, G. Animal models of chronic hepatitis delta virus infection host-virus immunologic interactions. Pathogens 2015, 4, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Amarán, L.; Usai, C.; Di Scala, M.; Godoy, C.; Ni, Y.; Hommel, M.; Palomo, L.; Segura, V.; Olagüe, C.; Vales, A.; et al. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-β induction. J. Hepatol. 2017, 67, 669–679. [Google Scholar] [CrossRef]
- Maestro, S.; Gómez-Echarte, N.; Camps, G.; Usai, C.; Suárez-Amarán, L.; Vales, A.; Olagüe, C.; Aldabe, R.; González-Aseguinolaza, G. AAV-HDV: An Attractive Platform for the In Vivo Study of HDV Biology and the Mechanism of Disease Pathogenesis. Viruses 2021, 13, 788. [Google Scholar] [CrossRef] [PubMed]
- Usai, C.; Maestro, S.; Camps, G.; Olague, C.; Suárez-Amaran, L.; Vales, A.; Aragon, T.; Hommel, M.; Aldabe, R.; Gonzalez-Aseguinolaza, G. TNF-alpha inhibition ameliorates HDV-induced liver damage in a mouse model of acute severe infection. JHEP Rep. 2020, 2, 100098. [Google Scholar] [CrossRef]
- Buitrago, B.; Popper, H.; Hadler, S.C.; Thung, S.N.; Gerber, M.A.; Purcell, R.H.; Maynard, J.E. Specific histologic features of Santa Marta hepatitis: A severe form of hepatitis d-virus infection in Northern South America. Hepatology 1986, 6, 1285–1291. [Google Scholar] [CrossRef]
- Verme, G.; Amoroso, P.; Lettieri, G.; Pierri, P.; David, E.; Sessa, F.; Rizzi, R.; Bonino, F.; Recchia, S.; Rizzetto, M. A histological study of hepatitis delta virus liver disease. Hepatology 1986, 6, 1303–1307. [Google Scholar] [CrossRef]
- Sagnelli, E.; Felaco, F.M.; Filippini, P.; Pasquale, G.; Peinetti, P.; Buonagurio, E.; Aprea, L.; Pulella, C.; Piccinino, F.; Giusti, G. Influence of HDV infection on clinical, biochemical and histological presentation of HBsAg positive chronic hepatitis. Liver 1989, 9, 229–234. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2013, 1, e00049. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Salvetti, A.; Pons, C.; Michelet, M.; Rivoire, M.; Baumert, T.F.; Durantel, D.; Lucifora, J. Loss of hepatitis D virus infectivity upon farnesyl transferase inhibitor treatment associates with increasing RNA editing rates revealed by a new RT-ddPCR method. Antiviral Res. 2022, 198, 105250. [Google Scholar] [CrossRef]
- Hwang, S.B.; Lai, M.M. Isoprenylation Masks a Conformational Epitope and Enhances trans-Dominant Inhibitory Function of the Large Hepatitis Delta Antigen. J. Virol. 1994, 68, 2958–2964. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Shih, K.; Lo, S.J. Ser-123 of the large antigen of hepatitis delta virus modulates its cellular localization to the nucleolus, SC-35 speckles or the cytoplasm. J. Gen. Virol. 2004, 85, 1685–1694. [Google Scholar] [CrossRef]
- Nakajima, T.; Nakashima, T.; Okada, Y.; Jo, M.; Nishikawa, T.; Mitsumoto, Y.; Katagishi, T.; Kimura, H.; Itoh, Y.; Kagawa, K.; et al. Nuclear size measurement is a simple method for the assessment of hepatocellular aging in non-alcoholic fatty liver disease: Comparison with telomere-specific quantitative FISH and p21 immunohistochemistry. Pathol. Int. 2010, 60, 175–183. [Google Scholar] [CrossRef]
- Lempp, F.A.; Schlund, F.; Rieble, L.; Nussbaum, L.; Link, C.; Zhang, Z.; Ni, Y.; Urban, S. Recapitulation of HDV infection in a fully permissive hepatoma cell line allows efficient drug evaluation. Nat. Commun. 2019, 10, 2265. [Google Scholar] [CrossRef]
- Cole, S.M.; Gowans, E.J.; Macnaughton, T.B.; Hall, P.M.; Burrell, C.J. Direct Evidence for Cytotoxicity Associated with Expression of Hepatitis Delta Virus Antigen. Hepatology 1991, 13, 845–851. [Google Scholar] [CrossRef]
- Lee, C.; Chen, P.; Lai, M.M.C.; Chen, D. Isoprenylation of large hepatitis delta antigen is necessary but not sufficient for hepatitis delta virus assembly. J. Virol. 1994, 199, 169–175. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NFκB. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C Virus Suppresses the IRE1-XBP1 Pathway of the Unfolded Protein Response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.H.; Ding, J.; Xie, X.X.; Yang, X.H.; Wu, X.F.; Chen, Z.X.; Guo, Q.L.; Gao, W.Y.; Wang, X.Z.; Li, D. Hepatitis B virus X protein promotes liver cell pyroptosis under oxidative stress through NLRP3 inflammasome activation. Inflamm. Res. 2020, 69, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Filipovska, J.; Yano, K.; Furuya, A.; Inukai, N.; Narita, T.; Wada, T.; Sugimoto, S.; Konarska, M.M.; Handa, H. Stimulation of RNA Polymerase II Elongation by Hepatitis Delta Antigen. Science 2001, 293, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Mura, T.; Chanarat, S.; Okamoto, S.; Handa, H. Hepatitis delta antigen binds to the clamp of RNA polymerase II and affects transcriptional fidelity. Genes Cells 2007, 12, 863–875. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDV Mutant | Primer | Sequence | Kit |
|---|---|---|---|
| HDV-NPrL-HDAg | Fw | 5′-TTTTCTCCCCAGAGTTCTCGACCCCAGTGAATAAAG-3′ | SDM (QuikChange II) |
| Rv | 5′-CTTTATTCACTGGGGTCGAGAACTCTGGGGAGAAAA-3′ | ||
| HDV-Ser177Ala | Fw 1 | 5′-ACTCCGGACCTGGGAAGAGGCCTCTCAGGGGAGGATTCAC-3′ | SDM (InFusion Cloning) |
| Rv 1 | 5′-TCCGAGAGAAGGGGGCCTCCGGGA-3′ | ||
| Fw 2 | 5′-AGGGAGTCCCGGAGGCCCCCTTCTCTC-3′ | ||
| Rv 2 | 5′-GAGCAGCGCTGCTCGAGGCAAGCTTGCATGCCTGCAGGTC-3′ | ||
| HDV-Ser177Asp | Fw 1 | 5′-ACTCCGGACCTGGGAAGAGGCCTCTCAGGGGAGGATTCAC-3′ | SDM (InFusion Cloning) |
| Rv 1 | 5′-GGTCCGAGAGAAGGGGTCCTCCGGGA-3′ | ||
| Fw 2 | 5′-AGGGAGTCCCGGAGGACCCCTTCTCTC-3′ | ||
| Rv 2 | 5′- GAGCAGCGCTGCTCGAGGCAAGCTTGCATGCCTGCAGGTC-3′ | ||
| HDV-ΔRibozyme | Fw 1 | 5′-CAAAGAATTGGGATTCGAACATCGATTGAATTCCCCGGGGA-3′ | SDM (InFusion Cloning) |
| Rv 1 | 5′-TCTTACCTGATGGGGCTCATGGTCCCA-3′ | ||
| Fw 2 | 5′-GAGGCTGGGACCATGAGCCCCATCAGGTAA-3′ | ||
| Rv 2 | 5′-AGCAAAGAAAGCAACGGGGCTAGCCGGTGGGTGTTC-3′ | ||
| HDAg-ΔNLS | Fw 1 | 5′-GGGGGCGGAACACCCACCGGCTAGCCCCGTTGCTTTCTTT-3′ | SDM (InFusion Cloning) |
| Rv 1 | 5′-TCGCCGGGGGAGCCCCTGCTCCATCCTTATCCTT-3′ | ||
| Fw 2 | 5′-AAGGATAAGGATGGAGCAGGGGCTCCCC-3′ | ||
| Rv 2 | 5′-GAGCAGCGCTGCTCGAGGCAAGCTTGCATGCCTGCAGGTC-3′ | ||
| HDAg-ΔNES | Fw 1 | 5′-ACTCCGGACCTGGGAAGAGGCCTCTCAGGGGAGGATTCAC-3′ | SDM (InFusion Cloning) |
| Rv 1 | 5′-TGGGGAGAAAAGGGTGCATCGGCT-3′ | ||
| Fw 2 | 5′-TCTTCCCAGCCGATGCACCCTTTTCT-3′ | ||
| Rv 2 | 5′-CTCTCGAGCAGCGCTGCTCGAGGCAAGCTT-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maestro, S.; Gomez-Echarte, N.; Camps, G.; Usai, C.; Olagüe, C.; Vales, A.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Deciphering the Role of Post-Translational Modifications and Cellular Location of Hepatitis Delta Virus (HDV) Antigens in HDV-Mediated Liver Damage in Mice. Viruses 2024, 16, 379. https://0-doi-org.brum.beds.ac.uk/10.3390/v16030379
Maestro S, Gomez-Echarte N, Camps G, Usai C, Olagüe C, Vales A, Aldabe R, Gonzalez-Aseguinolaza G. Deciphering the Role of Post-Translational Modifications and Cellular Location of Hepatitis Delta Virus (HDV) Antigens in HDV-Mediated Liver Damage in Mice. Viruses. 2024; 16(3):379. https://0-doi-org.brum.beds.ac.uk/10.3390/v16030379
Chicago/Turabian StyleMaestro, Sheila, Nahia Gomez-Echarte, Gracian Camps, Carla Usai, Cristina Olagüe, Africa Vales, Rafael Aldabe, and Gloria Gonzalez-Aseguinolaza. 2024. "Deciphering the Role of Post-Translational Modifications and Cellular Location of Hepatitis Delta Virus (HDV) Antigens in HDV-Mediated Liver Damage in Mice" Viruses 16, no. 3: 379. https://0-doi-org.brum.beds.ac.uk/10.3390/v16030379