
HIV-1 Capsid Rapidly Induces Long-Lived CPSF6 Puncta in Non-Dividing Cells, but Similar Puncta Already Exist in Uninfected T-Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Plasmids and Site-Directed Mutagenesis
2.3. Single-Round Infectious Virus Production
2.4. Immunofluorescence
2.5. Quantitative PCR Analysis to Measure RT Products
2.6. Statistics
3. Results
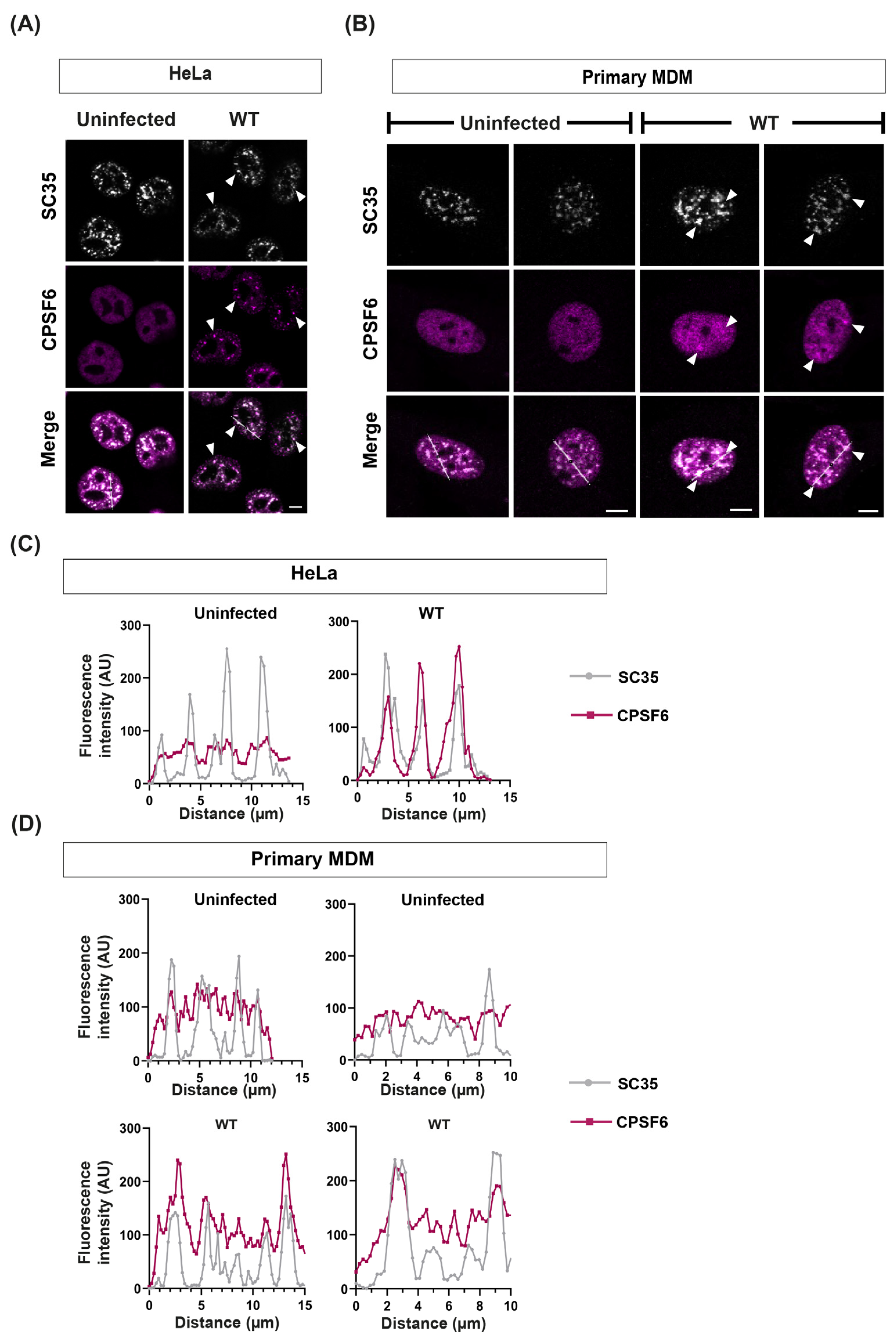
3.1. HIV Infection Relocalises CPSF6 into Puncta in Dividing and Non-Dividing Cells
3.2. CPSF5 but Not CPSF7 Is Relocalised with CPSF6
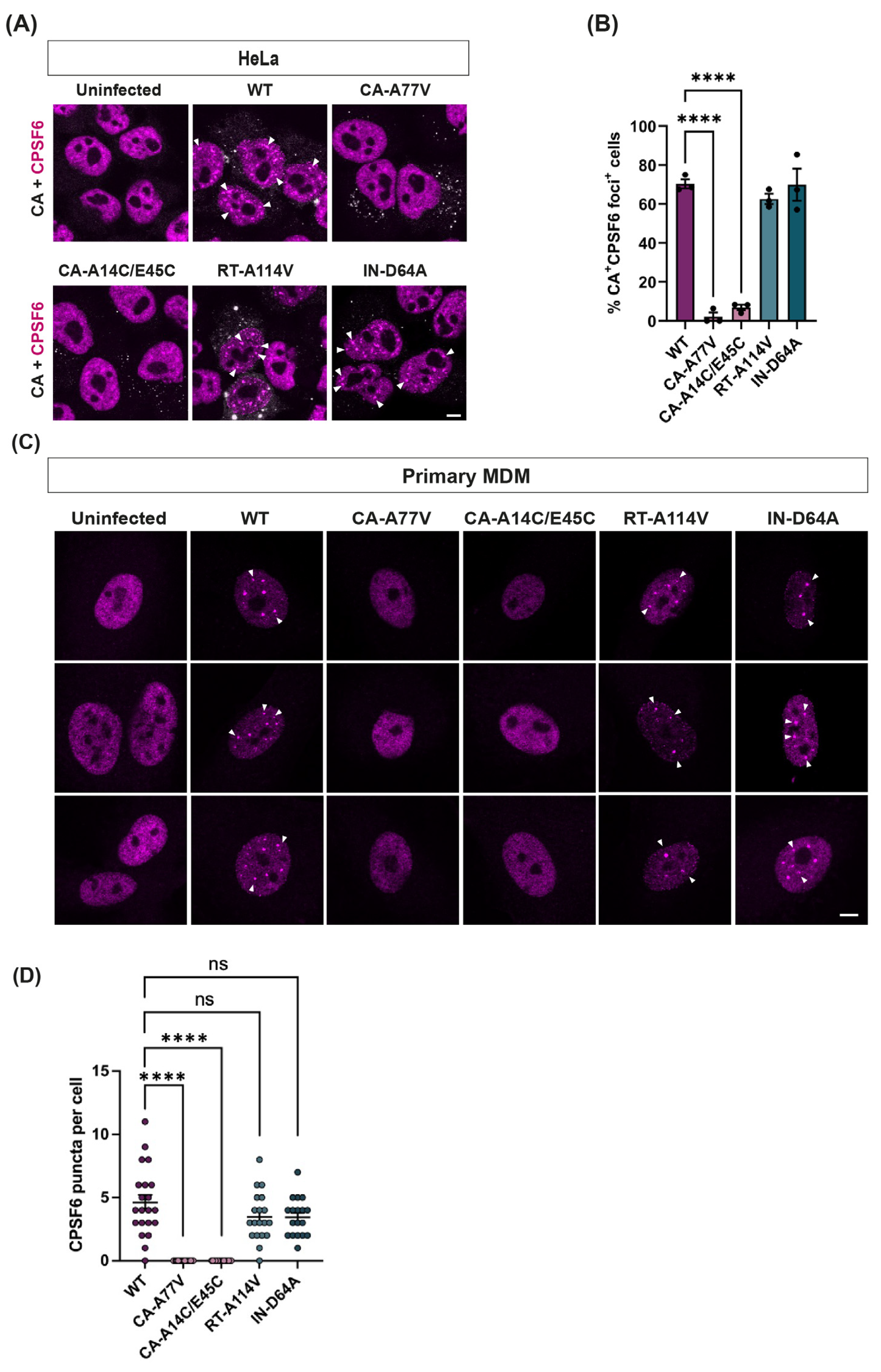
3.3. Reverse Transcription and Integration Are Not Required for Puncta Formation
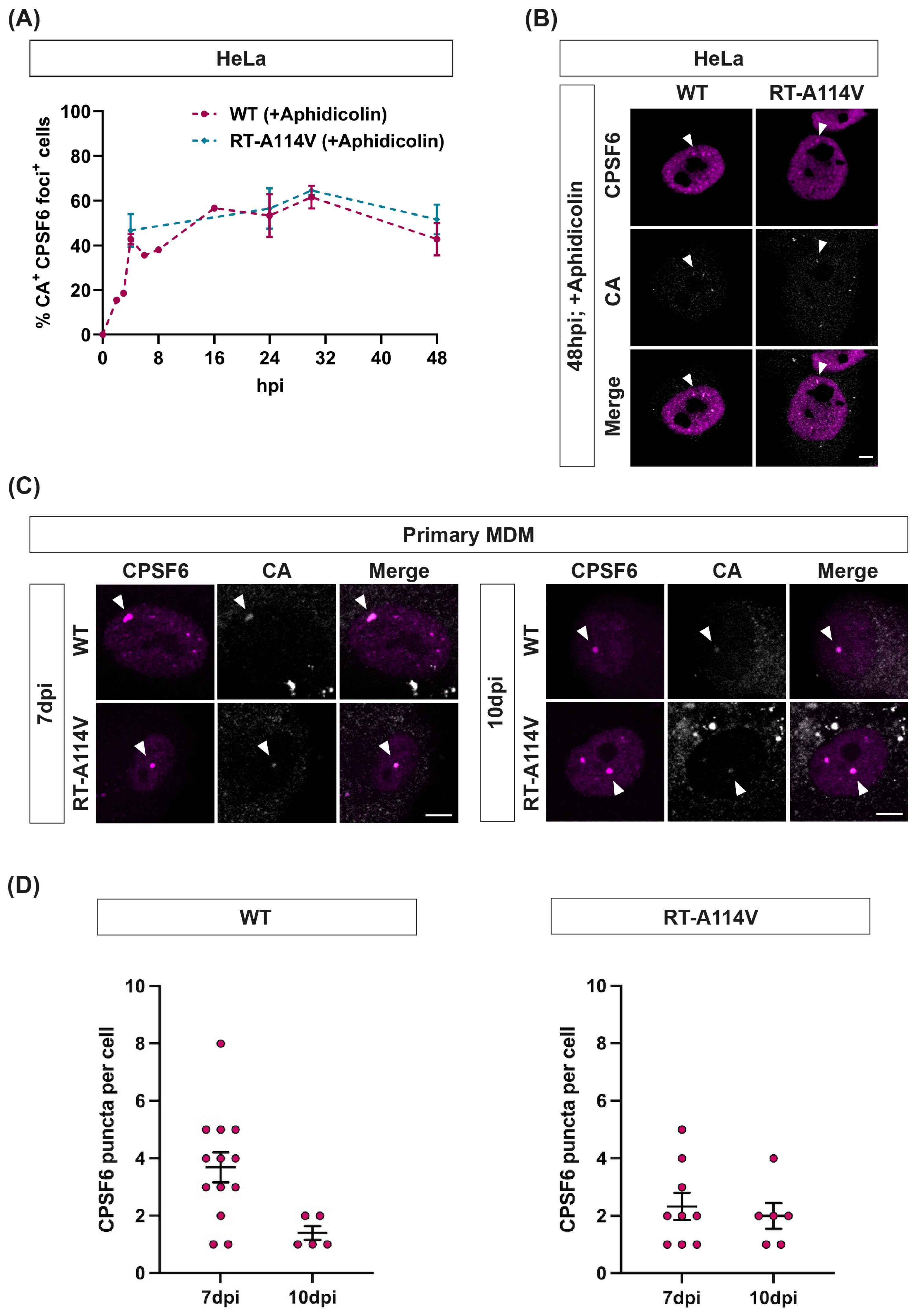
3.4. CPSF6 Puncta Form Rapidly and Decay Slowly in HeLa Cells
3.5. CPSF6 Puncta Form Rapidly and Can Exist for over Two Weeks in Macrophages
3.6. CPSF6 Puncta Remain until Cells Divide and Are Associated with CA Even at Late Times
3.7. The Number of Cells with Puncta Seems Proportional to the Amount of CA That Can Bind CPSF6
3.8. In Macrophages, the Number of Puncta Remain Constant Regardless of the MOI or the Number of Nuclear Speckles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gres, A.T.; Kirby, K.A.; KewalRamani, V.N.; Tanner, J.J.; Pornillos, O.; Sarafianos, S.G. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Glass, B.; Hagen, W.J.; Krausslich, H.G.; Briggs, J.A. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 2016, 354, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Guedan, A.; Caroe, E.R.; Barr, G.C.R.; Bishop, K.N. The Role of Capsid in HIV-1 Nuclear Entry. Viruses 2021, 13, 1425. [Google Scholar] [CrossRef] [PubMed]
- Cosnefroy, O.; Murray, P.J.; Bishop, K.N. HIV-1 capsid uncoating initiates after the first strand transfer of reverse transcription. Retrovirology 2016, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Hulme, A.E.; Perez, O.; Hope, T.J. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 9975–9980. [Google Scholar] [CrossRef] [PubMed]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, S.; Varadarajan, J.; Ramalho, R.; Aiken, C.; Rousso, I. Reverse Transcription Mechanically Initiates HIV-1 Capsid Disassembly. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fricke, T.; Diaz-Griffero, F. Inhibition of reverse transcriptase activity increases stability of the HIV-1 core. J. Virol. 2013, 87, 683–687. [Google Scholar] [CrossRef]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1088–1095. [Google Scholar] [CrossRef]
- Francis, A.C.; Marin, M.; Prellberg, M.J.; Palermino-Rowland, K.; Melikyan, G.B. HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines and in Primary Macrophages. Viruses 2020, 12, 1234. [Google Scholar] [CrossRef]
- Selyutina, A.; Persaud, M.; Lee, K.; KewalRamani, V.; Diaz-Griffero, F. Nuclear Import of the HIV-1 Core Precedes Reverse Transcription and Uncoating. Cell Rep. 2020, 32, 108201. [Google Scholar] [CrossRef] [PubMed]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef] [PubMed]
- Guedan, A.; Donaldson, C.D.; Caroe, E.R.; Cosnefroy, O.; Taylor, I.A.; Bishop, K.N. HIV-1 requires capsid remodelling at the nuclear pore for nuclear entry and integration. PLoS Pathog. 2021, 17, e1009484. [Google Scholar] [CrossRef] [PubMed]
- von Schwedler, U.K.; Stray, K.M.; Garrus, J.E.; Sundquist, W.I. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 2003, 77, 5439–5450. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Emerman, M. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J. Virol. 2004, 78, 5670–5678. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Perez, O.; Hope, T.J.; Emerman, M. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog. 2007, 3, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Borner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Muller, B.; Krausslich, H.G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. eLife 2019, 8, e41800. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Rebensburg, S.V.; Takata, M.A.; Zang, T.M.; Yamashita, M.; Kvaratskhelia, M.; Bieniasz, P.D. Nuclear pore heterogeneity influences HIV-1 infection and the antiviral activity of MX2. eLife 2018, 7, e35738. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Yucel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef]
- Shen, Q.; Feng, Q.; Wu, C.; Xiong, Q.; Tian, T.; Yuan, S.; Shi, J.; Bedwell, G.J.; Yang, R.; Aiken, C.; et al. Modeling HIV-1 nuclear entry with nucleoporin-gated DNA-origami channels. Nat. Struct. Mol. Biol. 2023, 30, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Kumari, S.; Xu, C.; Jang, S.; Shi, J.; Burdick, R.C.; Levintov, L.; Xiong, Q.; Wu, C.; Devarkar, S.C.; et al. The capsid lattice engages a bipartite NUP153 motif to mediate nuclear entry of HIV-1 cores. Proc. Natl. Acad. Sci. USA 2023, 120, e2202815120. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Machado, A.K.; Lyonnais, S.; Chamontin, C.; Gartner, K.; Leger, T.; Henriquet, C.; Garcia, C.; Portilho, D.M.; Pugniere, M.; et al. Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol. 2019, 4, 1840–1850. [Google Scholar] [CrossRef]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef]
- Tabasi, M.; Nombela, I.; Janssens, J.; Lahousse, A.P.; Christ, F.; Debyser, Z. Role of Transportin-SR2 in HIV-1 Nuclear Import. Viruses 2021, 13, 829. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ning, J.; Boggs, E.A.; Jang, S.; Wallace, C.; Telmer, C.; Bruchez, M.P.; Ahn, J.; Engelman, A.N.; Zhang, P.; et al. Cytoplasmic CPSF6 Regulates HIV-1 Capsid Trafficking and Infection in a Cyclophilin A-Dependent Manner. mBio 2021, 12, 10-1128. [Google Scholar] [CrossRef]
- Ay, S.; Di Nunzio, F. HIV-Induced CPSF6 Condensates. J. Mol. Biol. 2023, 435, 168094. [Google Scholar] [CrossRef]
- Hardy, J.G.; Norbury, C.J. Cleavage factor Im (CFIm) as a regulator of alternative polyadenylation. Biochem. Soc. Trans. 2016, 44, 1051–1057. [Google Scholar] [CrossRef]
- Ruegsegger, U.; Beyer, K.; Keller, W. Purification and characterization of human cleavage factor Im involved in the 3’ end processing of messenger RNA precursors. J. Biol. Chem. 1996, 271, 6107–6113. [Google Scholar] [CrossRef]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef]
- Francis, A.C.; Marin, M.; Singh, P.K.; Achuthan, V.; Prellberg, M.J.; Palermino-Rowland, K.; Lan, S.; Tedbury, P.R.; Sarafianos, S.G.; Engelman, A.N.; et al. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020, 11, 3505. [Google Scholar] [CrossRef] [PubMed]
- Rensen, E.; Mueller, F.; Scoca, V.; Parmar, J.J.; Souque, P.; Zimmer, C.; Di Nunzio, F. Clustering and reverse transcription of HIV-1 genomes in nuclear niches of macrophages. EMBO J. 2021, 40, e105247. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Li, P.; Yu, H.; Chen, L.L. Emerging roles of nuclear bodies in genome spatial organization. Trends Cell Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Singh, P.K.; Sowd, G.A.; Bedwell, G.J.; Jang, S.; Achuthan, V.; Oleru, A.V.; Wong, D.; Fadel, H.J.; Lee, K.; et al. CPSF6-Dependent Targeting of Speckle-Associated Domains Distinguishes Primate from Nonprimate Lentiviral Integration. mBio 2020, 11, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Rasheedi, S.; Shun, M.C.; Serrao, E.; Sowd, G.A.; Qian, J.; Hao, C.; Dasgupta, T.; Engelman, A.N.; Skowronski, J. The Cleavage and Polyadenylation Specificity Factor 6 (CPSF6) Subunit of the Capsid-recruited Pre-messenger RNA Cleavage Factor I (CFIm) Complex Mediates HIV-1 Integration into Genes. J. Biol. Chem. 2016, 291, 11809–11819. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, V.; Perreira, J.M.; Ahn, J.J.; Brass, A.L.; Engelman, A.N. Capsid-CPSF6 interaction: Master regulator of nuclear HIV-1 positioning and integration. J. Life Sci. 2019, 1, 39–45. [Google Scholar] [CrossRef]
- Scoca, V.; Morin, R.; Collard, M.; Tinevez, J.Y.; Di Nunzio, F. HIV-induced membraneless organelles orchestrate post-nuclear entry steps. J. Mol. Cell Biol. 2023, 14, mjac060. [Google Scholar] [CrossRef]
- Wei, G.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Singh, P.K.; Hudait, A.; Annamalai, A.S.; Bester, S.; Huang, S.W.; Shkriabai, N.; et al. Prion-like low complexity regions enable avid virus-host interactions during HIV-1 infection. Nat. Commun. 2022, 13, 5879. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Stephens, C.; Parsley, K.; Demaison, C.; Halfyard, A.; Thrasher, A.J.; Ali, R.R. In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector; efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene Ther. 2001, 8, 1665–1668. [Google Scholar] [CrossRef]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef]
- Li, W.; Lee, M.H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, J.; Naessens, E.; Vanderstraeten, H.; Landi, A.; Iannucci, V.; Van Nuffel, A.; Taghon, T.; Pizzato, M.; Verhasselt, B. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE 2012, 7, e50859. [Google Scholar] [CrossRef]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Saito, A.; Henning, M.S.; Serrao, E.; Dubose, B.N.; Teng, S.; Huang, J.; Li, X.; Saito, N.; Roy, S.P.; Siddiqui, M.A.; et al. Capsid-CPSF6 Interaction Is Dispensable for HIV-1 Replication in Primary Cells but Is Selected during Virus Passage In Vivo. J. Virol. 2016, 90, 6918–6935. [Google Scholar] [CrossRef] [PubMed]
- Van Cor-Hosmer, S.K.; Daddacha, W.; Kelly, Z.; Tsurumi, A.; Kennedy, E.M.; Kim, B. The impact of molecular manipulation in residue 114 of human immunodeficiency virus type-1 reverse transcriptase on dNTP substrate binding and viral replication. Virology 2012, 422, 393–401. [Google Scholar] [CrossRef]
- Faust, E.A.; Acel, A.; Udashkin, B.; Wainberg, M.A. Human immunodeficiency virus type 1 integrase stabilizes a linearized HIV-1 LTR plasmid in vivo. Biochem. Mol. Biol. Int. 1995, 36, 745–758. [Google Scholar] [PubMed]
- Diamond, T.L.; Roshal, M.; Jamburuthugoda, V.K.; Reynolds, H.M.; Merriam, A.R.; Lee, K.Y.; Balakrishnan, M.; Bambara, R.A.; Planelles, V.; Dewhurst, S.; et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 2004, 279, 51545–51553. [Google Scholar] [CrossRef]
- Towers, G.J.; Stockholm, D.; Labrousse-Najburg, V.; Carlier, F.; Danos, O.; Pages, J.C. One step screening of retroviral producer clones by real time quantitative PCR. J. Gene Med. 1999, 1, 352–359. [Google Scholar] [CrossRef]
- Trono, D. Partial reverse transcripts in virions from human immunodeficiency and murine leukemia viruses. J. Virol. 1992, 66, 4893–4900. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, C.; Lee, K.; Mardones, G.A.; KewalRamani, V.N.; Diaz-Griffero, F. Formation of nuclear CPSF6/CPSF5 biomolecular condensates upon HIV-1 entry into the nucleus is important for productive infection. Sci. Rep. 2023, 13, 10974. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, R.; Chen, L.; Deng, K.; Ou, X.; Lu, X.; Li, M.; Liu, C.; Chen, S.; Fu, Y.; et al. CPSF6 regulates alternative polyadenylation and proliferation of cancer cells through phase separation. Cell Rep. 2023, 42, 113197. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Cereseto, A.; Singh, P.K.; Shi, J.; Poeschla, E.; Engelman, A.N.; Aiken, C.; Melikyan, G.B. Localization and functions of native and eGFP-tagged capsid proteins in HIV-1 particles. PLoS Pathog. 2022, 18, e1010754. [Google Scholar] [CrossRef] [PubMed]
- Malikov, V.; da Silva, E.S.; Jovasevic, V.; Bennett, G.; de Souza Aranha Vieira, D.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6, 6660. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Muller, B.; Krausslich, H.G. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. eLife 2021, 10, e64776. [Google Scholar] [CrossRef]
- Bhat, P.; Chow, A.; Emert, B.; Ettlin, O.; Quinodoz, S.A.; Takei, Y.; Huang, W.; Blanco, M.R.; Guttman, M. 3D genome organization around nuclear speckles drives mRNA splicing efficiency. bioRxiv 2023. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guedán, A.; Burley, M.; Caroe, E.R.; Bishop, K.N. HIV-1 Capsid Rapidly Induces Long-Lived CPSF6 Puncta in Non-Dividing Cells, but Similar Puncta Already Exist in Uninfected T-Cells. Viruses 2024, 16, 670. https://0-doi-org.brum.beds.ac.uk/10.3390/v16050670
Guedán A, Burley M, Caroe ER, Bishop KN. HIV-1 Capsid Rapidly Induces Long-Lived CPSF6 Puncta in Non-Dividing Cells, but Similar Puncta Already Exist in Uninfected T-Cells. Viruses. 2024; 16(5):670. https://0-doi-org.brum.beds.ac.uk/10.3390/v16050670
Chicago/Turabian StyleGuedán, Anabel, Megan Burley, Eve R. Caroe, and Kate N. Bishop. 2024. "HIV-1 Capsid Rapidly Induces Long-Lived CPSF6 Puncta in Non-Dividing Cells, but Similar Puncta Already Exist in Uninfected T-Cells" Viruses 16, no. 5: 670. https://0-doi-org.brum.beds.ac.uk/10.3390/v16050670