Clinical Management of HIV Drug Resistance

HIV Drug Resistance Program, NCI, NIH, Building 10 Rm 5A06, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Viruses 2011, 3(4), 347-378; https://0-doi-org.brum.beds.ac.uk/10.3390/v3040347
Submission received: 9 March 2011
/
Accepted: 30 March 2011
/
Published: 14 April 2011
(This article belongs to the Special Issue HIV Drug Resistance 2010)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Combination antiretroviral therapy for HIV-1 infection has resulted in profound reductions in viremia and is associated with marked improvements in morbidity and mortality. Therapy is not curative, however, and prolonged therapy is complicated by drug toxicity and the emergence of drug resistance. Management of clinical drug resistance requires in depth evaluation, and includes extensive history, physical examination and laboratory studies. Appropriate use of resistance testing provides valuable information useful in constructing regimens for treatment-experienced individuals with viremia during therapy. This review outlines the emergence of drug resistance in vivo, and describes clinical evaluation and therapeutic options of the individual with rebound viremia during therapy.
1. Introduction
Combination antiretroviral therapy has resulted in marked improvements in morbidity and mortality from HIV-1 infection [1,2,3]. Therapy is not curative, however, and one of the most profound limitations of current antiretroviral therapy is the development of antiviral drug resistance [4]. HIV drug resistance occurs in a substantial proportion of treated patients and accumulates over time on therapy. Although the frequency of drug resistance has declined with the introduction of better tolerated regimens, resistance is still reported in 7–15% of patients initiating first line antiretroviral therapy [5,6,7]. Emergence of drug resistance has consequences for individuals and populations. For individuals, drug resistance restricts subsequent antiretroviral treatment choices and can exhaust therapeutic options, resulting in HIV-1 disease progression and death. Drug-resistant variants are also transmitted when new infections occur, effectively multiplying the individual drug failures and creating a growing public health concern. Surveillance studies report that the prevalence of drug-resistant mutations among recently infected drug-naïve individuals ranges from ca. 5–15% [8]. As such, transmission of drug resistance threatens to reverse the reductions in morbidity and mortality accomplished by antiretroviral therapy. Emergence of drug resistance is a consequence of a combination of viral, pharmacologic, and host factors. Identification, evaluation, and treatment of HIV drug resistance represent a compelling challenge for patients and health care professionals. Management of clinical resistance is a comprehensive process that determines the cause of rebound viremia and develops a useful course designed to re-suppress HIV replication. In this review we will describe factors in the development of drug resistance, and current issues in the clinical management of HIV drug resistance in vivo.
2. Sources of Drug Resistance
Since the development of antiretroviral therapy, over 20 chemotherapeutic agents have been FDA approved. Therapy is been directed against five principal viral targets, including attachment, fusion, reverse transcription, integration, and protease mediated maturation. Regardless of drug target, resistance may emerge to any antiretroviral, and viral, pharmacologic, and host factors contribute to the emergence of drug resistance. The replication program of HIV is rapid (T1/2 approximately 1 day) and error prone (mutation rate ca. 3 × 10−5 mutations/base/replication cycle) resulting in large and genetically diverse populations in vivo from which resistance may emerge [9]. Analysis of kinetics of emergence of drug resistance in vivo suggested that many single nucleotide mutations conferring drug resistance might be present prior to initiation of antiretroviral therapy. Early studies demonstrating rapid emergence of the single nucleotide mutations M184I and M184V resistance to 3TC suggested that therapy represents a selective pressure permitting emergence of resistant variants [10,11,12]. Pre-existing resistance is strongly supported by subsequent studies demonstrating the rapid, frequent emergence of drug resistance mutations after single dose of the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine [13]. Direct identification of drug resistance mutations pre-therapy has also been reported using sensitive allele-specific PCR amplification capable of detecting drug resistance at levels of ca. 0.3% [14].
Reverse transcriptase strand transfer events occur during reverse transcription. These events result in frequent recombination and as many as 6–7 strand transfers may take place during proviral synthesis. Reverse transcription and recombination of virions containing non-identical RNA copies encoding different individual resistance profiles will result in chimeric proviral DNA molecules with concatenated individual resistance mutations [15]. As a consequence, recombination is a potent mechanism for rapid spread of drug resistance mutations within an individual. Pharmacologic factors contribute to the development of resistance. In general, antiretroviral drugs are well absorbed and generate high drug levels capable of inhibiting HIV replication. Several agents, specifically the NNRTI class, have long half lives relative to the other regimen components. During non-adherent periods, short half life agents are eliminated relatively quickly, while longer agents become essentially monotherapeutic agents, which can select for drug resistance. Individuals taking antiretroviral medications often take additional therapeutic agents for co-morbid illnesses; drug interactions may result in changes in antiretroviral drug levels [16,17]. Although ongoing therapeutic drug monitoring has not become a clinic routine [18,19], drug level testing for all FDA approved antiretroviral is available and may be useful in evaluating whether sufficient drug levels are achieved, especially in individuals taking complex multidrug regimens to treat HIV and other illnesses, where drug interaction issues may arise.
Host factors, principally drug adherence, have a strong effect on the development of drug resistance. Early studies of antiretroviral therapy demonstrated frequent emergence of resistance to antiretroviral agents; regimens were complex, required frequent dosing, and were associated with a number of adverse effects; therapy interruption was relatively common, and rapidly resulted in development of drug resistance. With newer and better tolerated combination regimens, including those with once daily dosing, adherence generally improved in individuals taking first line regimens, and drug regimen failures have declined. Nevertheless, adherence remains a central issue in the development of resistance [3,20,21]. Direct observed therapy has been useful in investigating the virologic and immunologic effects of rigorously controlled drug delivery, although questions remain regarding the degree of improvement over voluntary therapy [22] and the content of care used to address adherence can predict virologic suppression [23]. Recent studies have suggested that the effect of nonadherence is not uniform [3], and that the probability of rebound viremia with non-adherence may decrease after viral suppression is achieved. A study from the REACH cohort studied 221 patients initiating antiretroviral therapy and estimated the probability of rebound viremia for various ranges of adherence after viral suppression is achieved. The probability of virologic failure after 1 month vs. 12 months of continuous HIV suppression with 50–74% adherence was 0.47, and 0.36 at 90–100% adherence [24]. In independent studies, Bello and colleagues [25] investigated the level of viremia associated with long-term suppression; viremia > 100 copies/mL plasma were associated with accumulation of new genetic diversity over time while little or no evidence was detected at lower viral RNA levels. The development of once daily regimens and combination formulation of antiretrovirals represent great improvements in therapy, making HIV therapy similar to therapy for other chronic diseases requiring daily therapy, such as therapy for hypertension, diabetes, and seizure disorders. Adherence to therapy for these other illnesses also presents a number of challenges; behavioral research in these diverse areas may yield new and useful strategies to improve adherence.
With the observations that HIV persists during therapy, it has become clear that understanding the nature of HIV replication during therapy has direct bearing on the potential for the emergence of drug resistance during therapy. If active spreading infection of HIV continues during drug suppression, then the potential for new mutations and drug resistance is possible. Alternatively, if drug suppression completely blocks spreading infection, the potential for emergence of new drug resistance mutations from chronically infected, long lived reservoirs is severely limited. Seminal studies by Persaud and coworkers demonstrated no emergence of new drug resistance mutations on therapy [26], while Martinez-Picardo and coworkers did identify emergence of mutations in individuals with prior therapy and transient viremic periods [27]. Subsequently, data supporting the presence and absence of active replication during therapy have been reported, reviewed by [28]. In a set of interventional studies, drug intensification has been used as a strategy to investigate whether ongoing replication takes place during suppressive antiretroviral therapy, reviewed by Maldarelli [29]; a number of studies have detected no evidence of decreased viremia during drug intensification using sensitive single copy detection assays. Buzon and colleagues [30] have identified patients with increased levels of 2-LTR circular DNA during raltegravir intensification, suggesting that, in some patients, residual HIV replication may be present. Research to confirm these findings and to characterize patients undergoing antiretroviral therapy with complete suppression or ongoing replication continues.
Host genetic variation can have strong effects on the course of HIV infection. There are strong host encoded differences in the rate of metabolism of antiretrovirals; such pharmacogenomic issues can contribute to drug half life, and may result in underexposure leading to resistance, or over-exposure leading to toxicity of antiretrovirals [31]. In addition, specific host traits, such as HLA, have direct effect on development of cutaneous hypersensitivity to drugs including the antiretroviral abacavir [32]. Research in this areas continues to expand to include genome wide understanding of host-virus interactions.
2.1. Laboratory methods for detection of antiviral drug resistance
2.1.1. Genotyping
Genotyping identifies resistance by a three step process of (1) nucleic acid analysis of relevant portions of the HIV genome derived from plasma, (2) identifying mutations associated with drug resistance, (3) constructing a drug resistance report. Nucleic acid analysis consists of extraction of HIV RNA from plasma, reverse transcription and PCR amplification of relevant portions of the HIV genome. HIV has broad genetic diversity that is lost, to a significant degree, during the amplification process; as a consequence only the most common variants (present in at least 15–20%) are represented in the genotypic information provided to the health care professional and the patient. Resistance associated mutations are typically “archived” in cells for apparently indefinite periods. Thus, it is possible that drug resistance may be present in the individual with rebound viremia on therapy but not detectable in resistance assays. Direct nucleic acid sequencing represents a common mechanism to obtain resistance information; commercial genotyping services, as well as systems for laboratory use are available; routine testing with independent panels of resistant viruses is useful to maintaining proficiency in detection of mutations [33,34,35]. Assays for specific mutations by selective hybridization are also available commercially in selected parts of the world; such “line probe” assays are reported to be more sensitive for detection of low level mutations, but are limited by the number of mutations available for assay [36,37,38]. It is possible that natural genetic variation surrounding the mutation site may affect detection of mutations. Mutations are identified by comparison with HIV “wild type” sequences. In the setting of wide genetic diversity “wild type” HIV is certainly only an approximation; commercially, laboratory infectious clones, such as pNL4-3 [39] or LAV [40] are used as representative viruses. Drug resistance sites have been identified in vivo and in vitro. In vivo, the emergence of specific changes on rebound viremia during therapy are identified and studied in vitro, demonstrating that introduction of specific mutations into wild type (WT) virus recapitulates drug resistance. In vitro cultivation studies also identify drug resistance mutations, typically with gradual emergence of resistance during long-term passage of HIV in the presence of antiretrovirals. The combination of in vivo and in vitro studies has yielded a comprehensive list of drug resistance mutations [35]. Commercial genotyping report provides the list of mutations conferring drug resistance, and also synthesizes the genotypic information into an interpretation provided to the health care professional, reporting predicted resistance for each drug. The predictions are either rule based, or the product of algorithms derived by analyses of large proprietary databases; all are detailed, but none are perfect [41,42]. The algorithms and rules are regularly updated with new accumulated data; new mutations may be added while others may be dropped. In addition, a number of public access sites (e.g., Stanford University HIV Drug Resistance Database [43]) are available to analyze genotypic information obtained from home-brew or commercially obtained sequences. Such sites may be useful for updated analysis of genotypes obtained 5–10 years ago that were originally interpreted with older algorithms.
In addition to reporting resistance identification, HIV drug resistance reports offer a wealth of additional useful information, particularly when combined with clinical information; five examples illustrate clinical utility of careful evaluation of genotypic data. First and most obviously, the absence of any drug resistance mutations, in the setting of high rebound viremia strongly suggests adherence issues may be the cause of rebound viremia. Second, the emergence or loss of certain drug resistance mutations responds relatively rapidly to the presence of the antiretroviral drug pressure. Resistance to 3TC or FTC is typically accompanied by the mutation M184V; M184I also confers resistance to cytidine analogues, but is usually only transiently detected, if at all, early during therapy with 3TC or FTC. M184I is rapidly outcompeted by the M184V mutation. As a result, the presence of the M184I mutation suggests that individuals have recently initiated (or re-initiated) therapy with 3TC. M184V itself responds relatively rapidly to the presence of 3TC; loss of this mutation during therapy with 3TC raises adherence concerns. Third, the presence of polymorphisms at resistance sites may be a useful observation. The presence of polymorphisms at sites strongly selected by the regimen (e.g., M41M/L, during AZT therapy) suggests that drug selection pressure has not been uniform, and non-adherence may play a role in rebound viremia. Fourth, genetic analysis and detecting the presence of uncommon mutations may offer useful insight into the origin of the infection in an individual. Genotypic analysis is used to determine HIV subtype; non-B subtypes are readily detected by this method, and can provide useful information regarding geographic source of infection. Uncommon mutations at position 215 of reverse transcriptase, such as T215S, T215C, reflect back mutations [44], suggest the prior presence of T215Y or F, and often represent strong evidence of transmitted drug resistance. Finally, some resistance sites are reported to confer resistance to one antiretroviral and may confer increased sensitivity to other antiretroviral drugs. Examples include K65R, which confers high level resistance to tenofovir, confers increased sensitivity to AZT; the mutation T215Y confers resistance to AZT, but is more sensitive to tenofovir than the wild type T215. Clinical correlates of these in vitro observations are uncertain but may be useful in constructing new regimens to treat the resistant virus [45].
2.1.2. Phenotyping
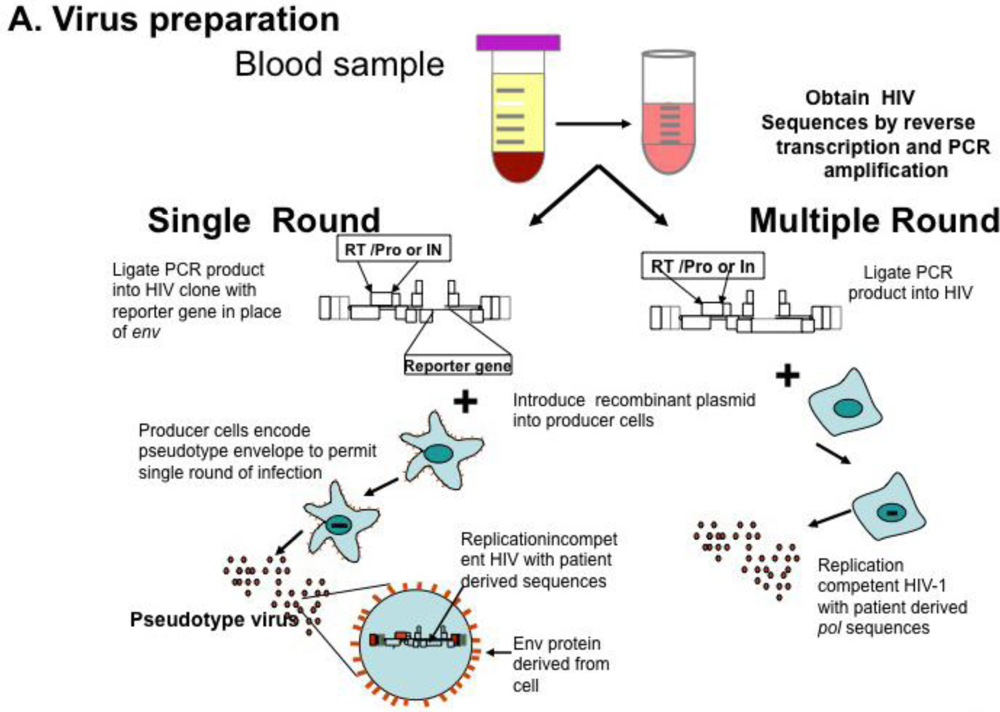
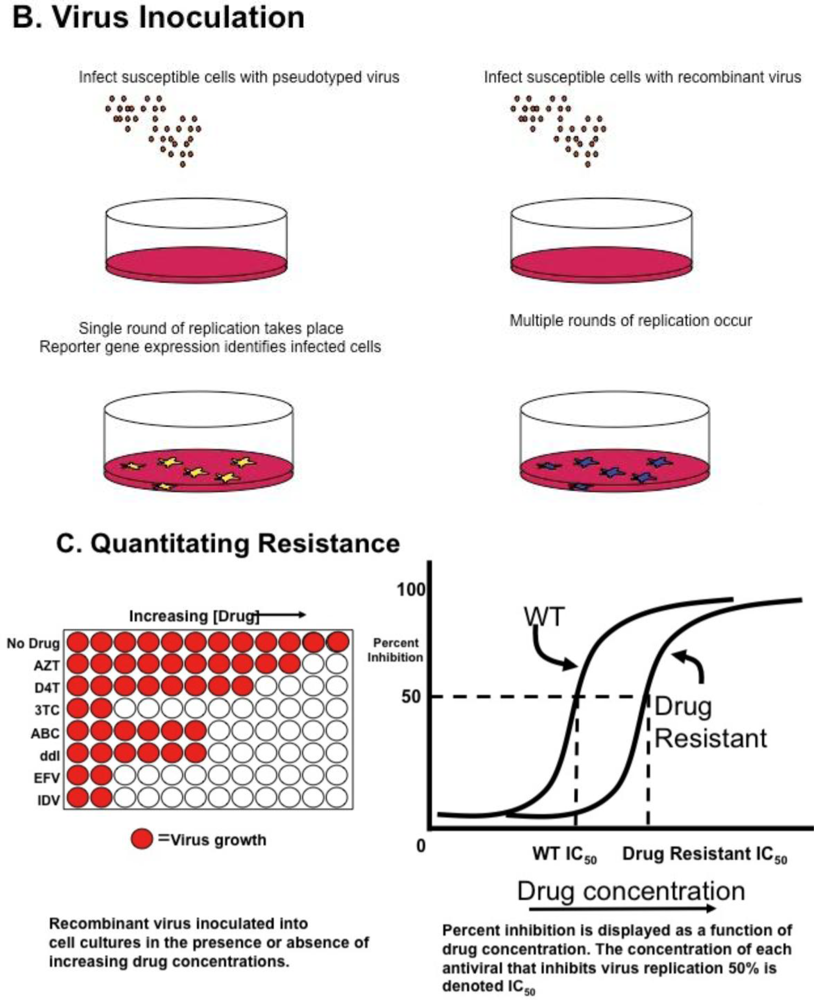
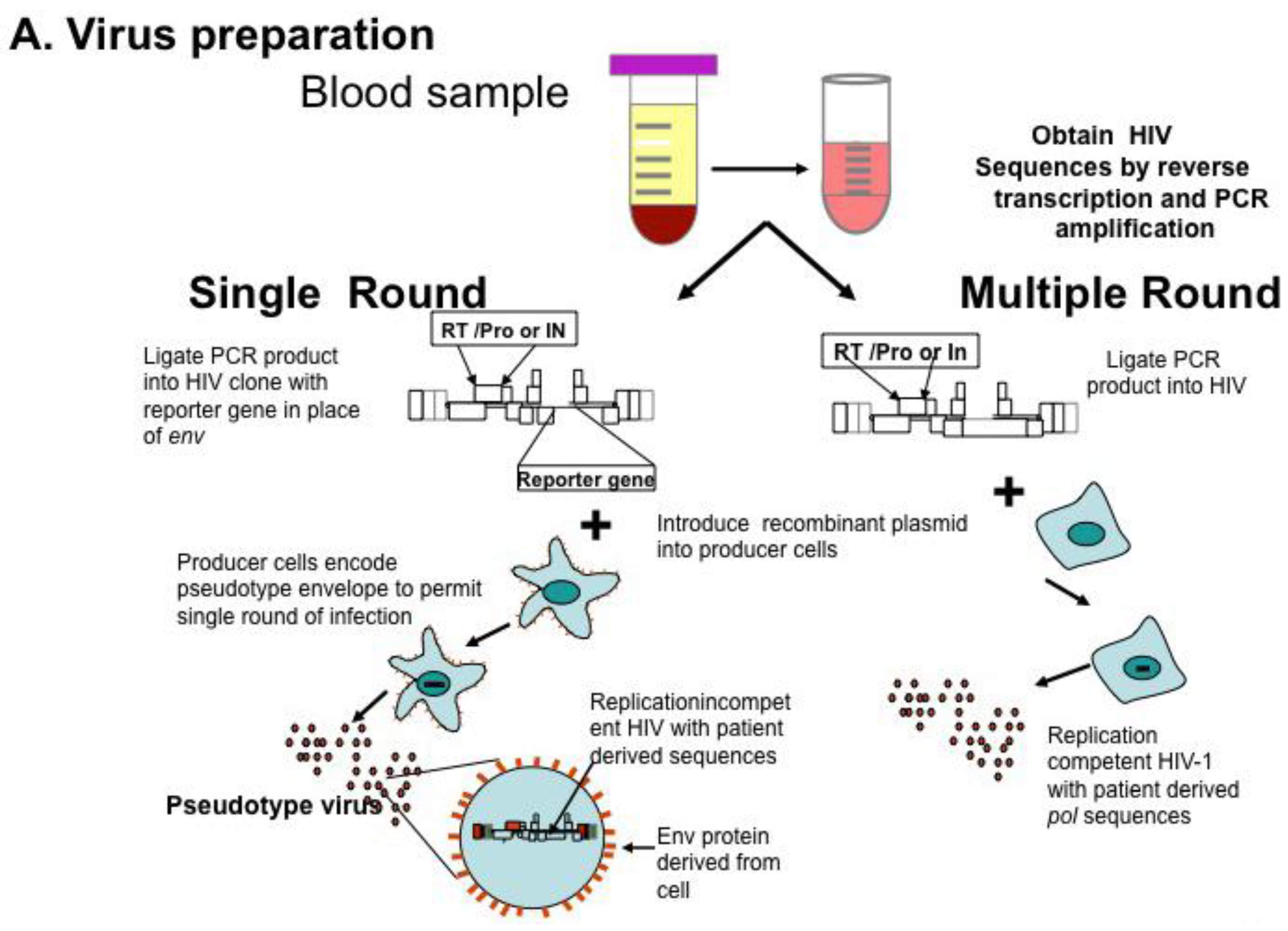
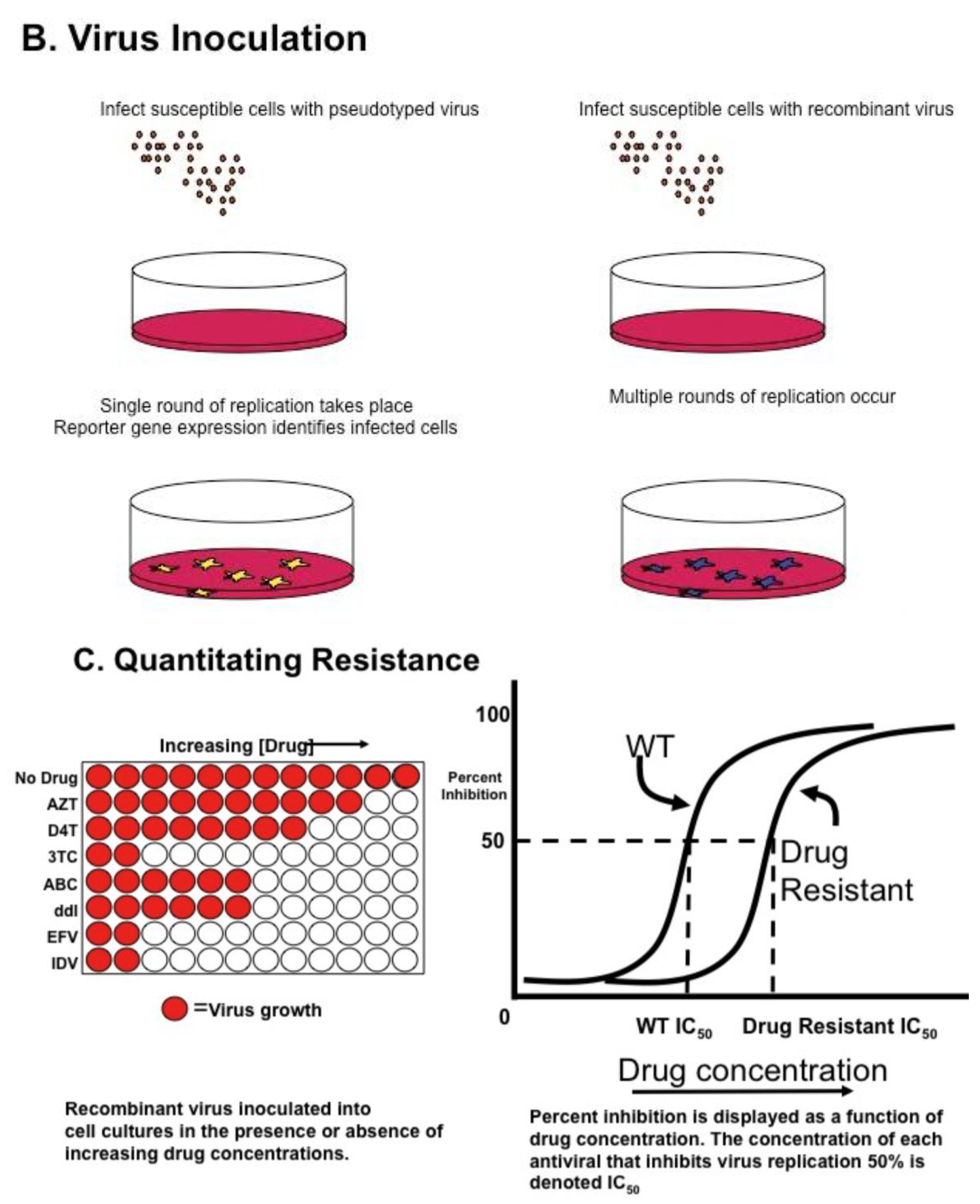
Genotyping information may yield complex mutational patterns. Phenotyping assays have been developed in an effort to provide a functional evaluation of patient derived HIV-1 protease and reverse transcriptase. Recombinant DNA technology [46] generating chimeric plasmids with gag/pol sequences from patient isolates cloned into laboratory-adapted HIV-1 strains permits reliable and reproducible measurement of in vitro resistance; several assays are commonly available using different strategies as single round or multiple round infections [35,47,48] (Figure 1A). HIV-1 protease, RT, integrase, and envelope sequences amplified from patient material using RT-PCR techniques as described above for genotyping are introduced into a recombinant molecular clone of HIV-1, either by direct ligation, or by simply mixing and allowing ligation to occur during the transfection [49]. Virions produced by transfection are standardized and used to inoculate cultures of susceptible cells (Figure 1B); in parallel, cultures are inoculated with wild type virus. Infections are carried out in the presence of increasing concentrations of individual antiviral agents, viral replication measured and dose response curves constructed (see Figure 1C); the concentration of drug necessary for 50% inhibition of virus replication is reported as IC50. Conceptually, the two approaches appear to offer different advantages. Single round assays eliminate the possibility that resistance may arise during the cultivation of wild type virus in the presence of the drug [50]. Multi-round cultivation assays may detect low level resistance that may not manifest in a single round assay. Direct comparison of these two assays revealed remarkably concordant results [51].
Both phenotyping systems evaluate only a portion of patient derived material; any interactions between portions of HIV genome from patient derived virus are not measured. For example, phenotypic analysis of patient derived HIV protease takes place without inclusion of most protease cleavage sites as additional compensatory changes may occur at cleavage sites, it is likely that a degree of phenotypic information is lost in the process [52,53,54]. New phenotypic assays have been developed to investigate HIV tropism to evaluate patients who are under consideration for therapy with coreceptor inhibitors. Such assays are can detect the presence of X4 or dual X4/R5 tropic virus at low levels and are recommended prior to drug initiation, as the presence of X4 or dualtropic virus is likely not to be susceptible to coreceptor inhibitors in viremic patients [35].
Increases in IC50 are associated with drug resistance, but the degree of virologic resistance associated with clinical drug failure is not clear in all circumstances. Establishment of effective cutoffs has been a major effort in phenotyping development. For some antiretrovirals, such as efavirenz or 3TC, large increases in IC50 are noted, and identifying resistance is straightforward. In contrast, drugs such as ddI or d4T have a more restricted dynamic range in phenotyping assays. Phenotypic sensitivity scores were predictive of viral RNA responses in treatment experienced patients initiating protease inhibitor regimens [55], demonstrating the utility of phenotypic results and RNA responses, but precise cutoffs are relatively difficult to assign. One approach has been to evaluate the distribution of drug resistance in isolates from drug naïve individuals and assigning cutoffs at IC50 levels several standard deviations beyond the mean drug naïve level [56]. Use of clinical trial and cohort analysis has also been proposed to estimate clinical cutoffs [57]. Increasing knowledge regarding the nature of drug resistance and the success of commercial drug resistance testing has permitted analysis of large numbers of matched HIV genotypes and phenotypes and development of bioinformatics tools that predict phenotypic responses based solely on genotypic information. These analyses have been studied in clinical trials and are commercially available [58,59,60] or open access [61] e.g., [62]or ANRS [63] and Rega algorithms [64] and the Stanford HIVDB [43] are generally well correlated with experimental phenotypic drug resistance data and have recently been reviewed [65,66]. A current list of resistance mutations is maintained and updated regularly [67].
Figure 1.
HIV Phenotyping of HIV protease (PRO), RT, integrase (IN) in Cell Based Assays. (A) Schematic of sample processing from phlebotomy to construction of chimeric recombinant plasmids composed of patient derived sequences in standard laboratory based HIV standard clones. Single round assays use HIV derivatives encoding a reporter gene instead of HIV env. Upon transfection into producer cells expressing a helper virus envelope, virions are produced which can undergo a single round of replication. Multiple round assays introduce patient-derived material into standard laboratory HIV, and recombinant plasmids transfected into producer cells. (B) Virions produced by transfection are standardized and used to infect susceptible cells. In single round assays, pseudo typed viruses undergo reverse transcription and integration, but are unable to propagate. Production of reporter gene product (e.g., luciferase, green fluorescent protein) denotes successful round of replication. In multiple round infections, virus is inoculated and production measured by standardized measures, typically production of p24 antigen in media. (C) To determine phenotypic response to antivirals, virus is inoculated in the presence of increasing concentrations of single antiretrovirals. Dose response curves are constructed and measure of drug inhibition; the amount of drug necessary to inhibit 50% of virus replication (IC50) is calculated. Adapted from [68].
Figure 1.
HIV Phenotyping of HIV protease (PRO), RT, integrase (IN) in Cell Based Assays. (A) Schematic of sample processing from phlebotomy to construction of chimeric recombinant plasmids composed of patient derived sequences in standard laboratory based HIV standard clones. Single round assays use HIV derivatives encoding a reporter gene instead of HIV env. Upon transfection into producer cells expressing a helper virus envelope, virions are produced which can undergo a single round of replication. Multiple round assays introduce patient-derived material into standard laboratory HIV, and recombinant plasmids transfected into producer cells. (B) Virions produced by transfection are standardized and used to infect susceptible cells. In single round assays, pseudo typed viruses undergo reverse transcription and integration, but are unable to propagate. Production of reporter gene product (e.g., luciferase, green fluorescent protein) denotes successful round of replication. In multiple round infections, virus is inoculated and production measured by standardized measures, typically production of p24 antigen in media. (C) To determine phenotypic response to antivirals, virus is inoculated in the presence of increasing concentrations of single antiretrovirals. Dose response curves are constructed and measure of drug inhibition; the amount of drug necessary to inhibit 50% of virus replication (IC50) is calculated. Adapted from [68].


Direct comparisons of virtual phenotypic data with genotype and phenotype have not been extensively investigated. In the CREST trial, Emrey and colleagues [69] did not detect a difference in virologic success in a randomized study of individuals undergoing drug resistance testing with or without virtual phenotyping; Saracino and coworkers obtained similar virologic suppression in patients using virtual or real phenotypic results [59]. A number of research approaches have incorporated IC50 phenotypic data with levels of drug measured in individual patients. These inhibitory quotients have correlated well with drug suppression [70,71], but have not been used extensively yet in routine clinical practice.
An entirely new analysis of phenotypic data derives new and useful information from slopes of the dose response curve [72] used to determine instantaneous inhibitory potentials. Dose response slopes were found to be class specific, and these authors established limits for the inhibitory activity of individual drugs and for entire classes. In this analysis, NNRTIs and protease inhibitors had the greatest activity against wild type virus. The performance characteristics of the instantaneous inhibitory potential have not yet been field tested in detail or with drug resistant virus. An initial analysis by Kuritzkes and colleagues obtained similar outcome data using IIP or IQ, and the clinical utility of these modalities remains under study [73].
3. Clinical Utility of Drug Resistance Testing
3.1. Evidence Base for the Use of Drug Resistance Assays in Clinical Management of HIV-1 Infection
A number of randomized, prospective studies investigated the utility of resistance testing in management of antiretrovirals [74,75,76,77,78]. VIRADAPT [79] and GART [80] were two early prospective genotyping studies that randomized patients to two arms: genotyping and standard of care. The benefit in both of these trials appeared to be the ability of genotyping to identify a greater number of active antiretrovirals. VIRA3001 [75] was a randomized study comparing phenotyping and standard of care in 272 viremic patients; at the week 16 endpoint phenotyping arm had demonstrated a greater effect on reducing HIV-1 viremia measured by proportion of patients <400 c/mL; in the secondary analysis, phenotyping was also superior to SOC in reducing viremia, measured by area under the curve minus baseline. The specific benefit of additional “expert advice” e.g., panels of investigators with extensive experience in HIV therapy was specifically investigated in HAVANA [78], a factorial design study comparing both the use of genotyping and the use of expert advice in virologic outcome (VL < 400 c/mL). Genotyping was superior to no genotyping even in the absence of expert advice, using only the algorithm interpretation of the genotype. In addition, expert advice was beneficial even in the absence of genotyping; more recently, higher agreement rates among those with “expert opinion” suggests more consensus among therapeutic options [81].
Duration of these studies were relatively short; ARGENTA was a prospective randomized study comparing efficacy of genotyping and standard of care at two time points [76] in reducing viral loads. Although the proportion of patients suppressed <500 c/mL was significantly greater in the genotyping arm after 12 weeks, the effect was not sustained and no benefit of genotyping was detected at 24 weeks. The durability of resistance testing beyond one year has not been studied prospectively. Many of the studies were performed prior to widespread use of sensitive <50 copy limit assays and used <400 c/mL or <500 c/mL as a measure of success; it is not clear whether all successful treatments reached the more stringent measure of suppression, but the virologic benefit realized in genotyping arm of the ARGENTA was persistent in subsequent longitudinal analysis [76].
Several trials have not demonstrated benefit of resistance testing. NARVAL was a randomized study of phenotyping, genotyping and standard of care [77] no benefit of resistance testing was noted in the entire cohort; in a sub analysis of patients a benefit of genotyping in patients with a single prior PI-containing regimen was detected. NARVAL patients were in general more drug experienced than in prior trials, and the absence of effective drugs regardless of resistance testing may have contributed to the equal success rates. NARVAL was initiated after resistance testing had been generally introduced, and physicians had the benefit of earlier trials of the performance of antivirals in various drug resistance settings.
In these early studies, participants were generally less antiviral experienced than patients currently failing therapy. The clinical benefit of resistance testing in viral suppression in highly experienced patients has not been extensively investigated. In NARVAL [77], no significant differences in adverse event reporting were detected in resistance testing and standard of care arms. The CCTG575 Study of 238 patients with drug resistance did not demonstrate a clear benefit of phenotyping over standard of care, although benefits were noted in patients with more resistant virus [82]. A meta analysis of the effectiveness of drug resistance testing of ten trials highlighted the short term nature of the benefits of genotyping and virtual phenotyping [83]. In contrast, retrospective analysis of 2699 of the HOPS natural history cohort study patients over the period 1999–2005 revealed a survival advantage in management of drug resistance that included drug resistance testing compared with expert advice alone [84]. Drug resistance testing has become part of the standard of care of individuals with rebound viremia and remains recommended by guidelines for treatment of HIV infected individuals [85]. Resistance testing is recommended in the initial evaluation of HIV–infected individuals at the time of diagnosis, in order to identify individuals with transmitted drug resistance and at confirmed rebound in viremia [85,86].
4. Drug Class Specific Issues
4.1. NRTI
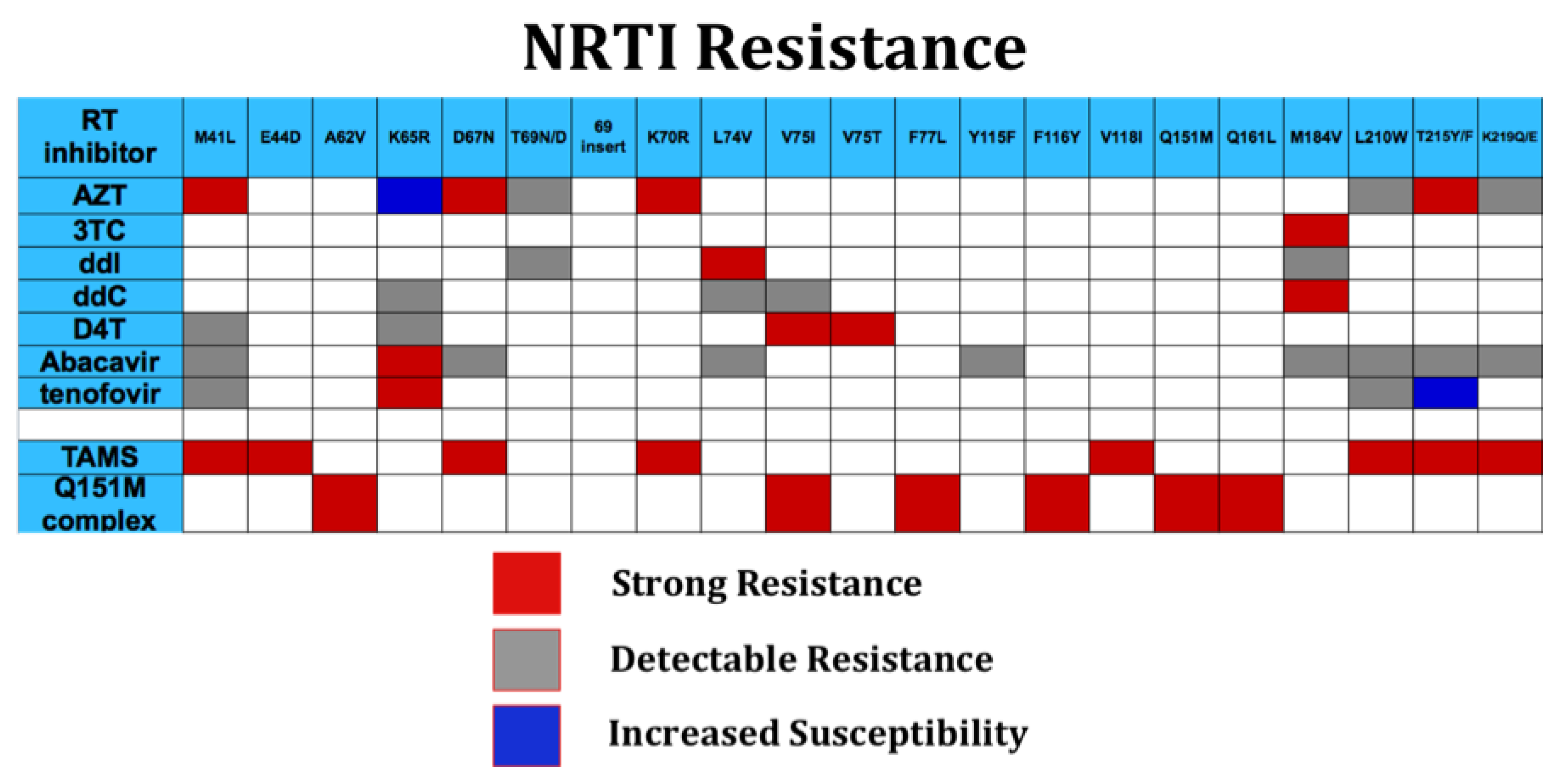
Reverse transcriptase is the central enzyme in HIV replication and mediates RNA-dependent DNA synthesis, RNase H excision of HIV RNA from RNA: DNA hybrid, and DNA directed DNA synthesis. A reverse, “excision” reaction, which removes incorporated nucleotides, also occurs at a low rate compared to polymerization. RT assembles as a dimer of p66 and p51 subunits; like a number of DNA polymerases, the structure of RT is similar to a right hand, and includes a palm, fingers, and thumb domains [87]. Nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs) represent strong inhibitors of reverse transcriptase and inhibit polymerization by chain termination. NRTIs such as AZT, 3TC, and ddI represent some of the oldest antiretrovirals, and many individuals with long antiretroviral experience have had extensive exposure to NRTIs singly or in combination, with accumulation of a number of drug resistance mutations (Figure 2). In general, mutations, result in steric effects on drug access, e.g., M184V, 3TC, ddC [88], or kinetic effects that result in marked increase in excision of incorporated analogues [89]; thymidine associated mutations (TAMs, including M41L, D67N, K70R, L210W, T215Y/F, K219Q) confer resistance by enhanced excision. TAMs excision may be reduced by M184V. In addition, M184V-containing virus is less fit than wild type virus [90,91], perhaps providing an additional virologic benefit. Accumulation of TAMS results in increasing resistance to AZT, tenofovir, D4T, abacavir, and ddI. Two multidrug resistance mutation profiles in general, confer high level drug resistance to all NRTIs. The Q151M suite of mutations (Figure 2) results in selective decreased binding of NRTI triphosphate and a degree of increased excision [92,93] ; although tenofovir has some reported activity in this setting [94], while other NRTIs such as AZT, D4T, ddI, and abacavir are uniformly ineffective. Insertions following the threonine residue at position 69, typically consisting of 1–4 additional residues result in uniformly high level resistance to all NRTIs.
Accumulation of drug resistance mutations reduces the rate of reverse transcription. RNase H activity of RT, which removes RNA from the RNA:DNA product of first round reverse transcription must work in concert with polymerization. Pathak and coworkers hypothesized that additional mutations affecting RNase H activity may compensate for accumulated mutations slowing RT mediated polymerization [95]. Such resistance mutations have been identified in the connection domain of reverse transcriptase [95,96,97]. Although this region of RT is not typically included in commercial genotyping, connection domain mutations may increase resistance to certain NRTIs, such as AZT, by 10–100 fold [35,96,97]. The clinical consequences of these new NRTI resistance mutations remain under intense study.
4.2. NNRTI
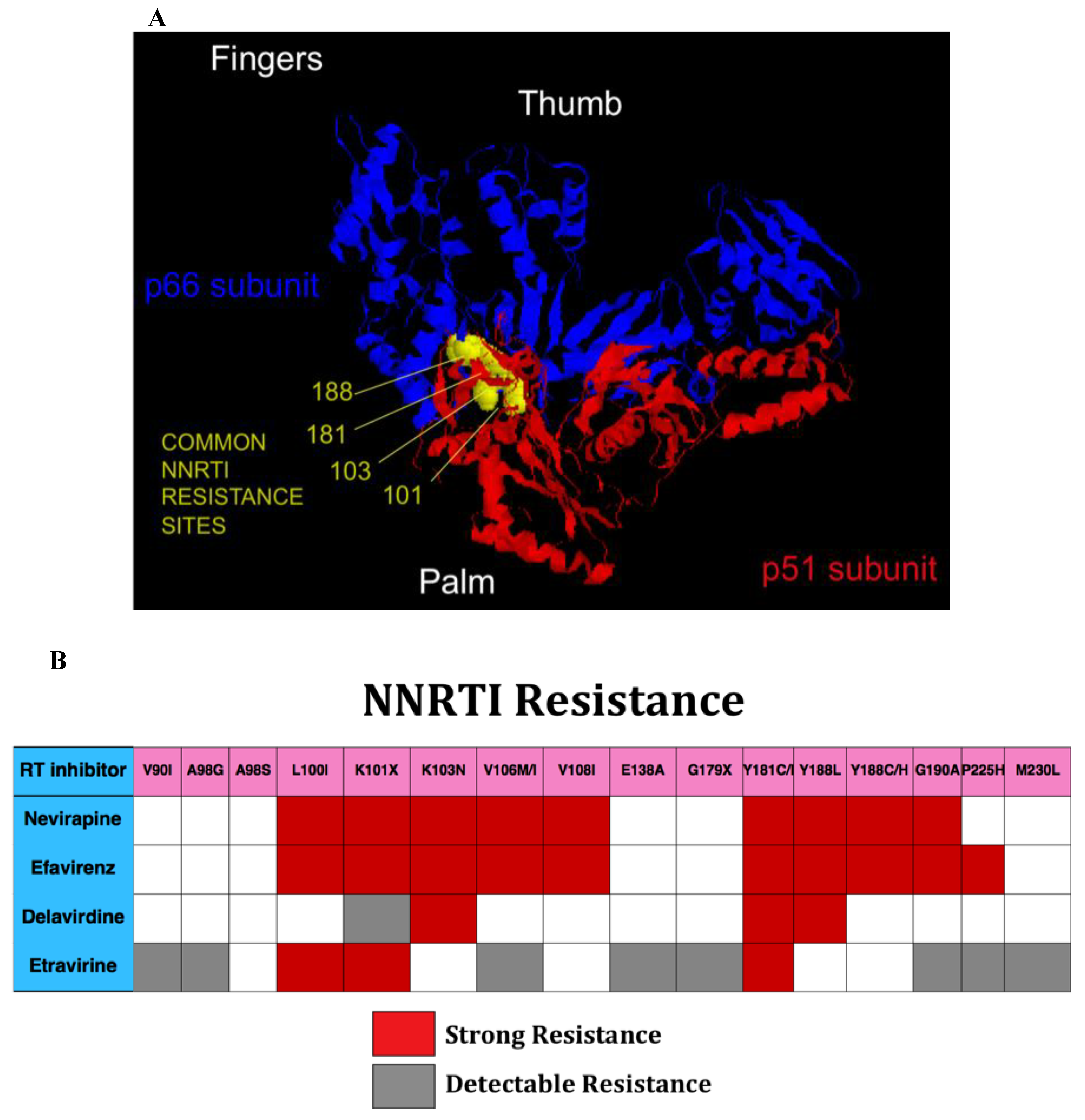
NNRTIs bind to a common hydrophobic pocket in the palm domain of RT that is near, but not at the HIV RT catalytic site (Figure 3A). Mutations conferring resistance result in changes in binding characteristics for the inhibitors [98]. In general, there is extensive cross-resistance among NNRTIs (Figure 3B). The majority of NNRTI resistance mutations do not significantly reduce the replication capacity of HIV, and tend to persist for prolonged periods. The newest NNRTI, etravirine, has significant antiviral activity even in the setting of a number of NNRTI resistance mutations, including K103N and Y188C. As a result it is possible to construct NNRTI-based regimens in individuals with NNRTI resistance. One challenging aspect of such a strategy is ensuring that etravirine mutations (Y181C) are not present in individuals, some of whom may have remotely taken NNRTI. It is not clear that relying on population-based based genotypes, which only report on contemporary and predominant HIV sequences, will provide sufficient sensitivity to rule out the presence of concerning mutations.
Several studies have demonstrated new RT mutations accumulating on NRTI that confer increased susceptibility to reverse transcriptase [102,103,104]. The mechanism by which new mutations confer hypersusceptibility to NNRTI is uncertain. Hypersusceptibility may affect nucleotide selectivity or virus replication capacity [105,106]. Clinical advantage of NNRTI hypersusceptibility has not been extensively investigated, but patients with hypersusceptibility did experience higher reductions in viral RNA, suggesting a clinical advantage [103,107].
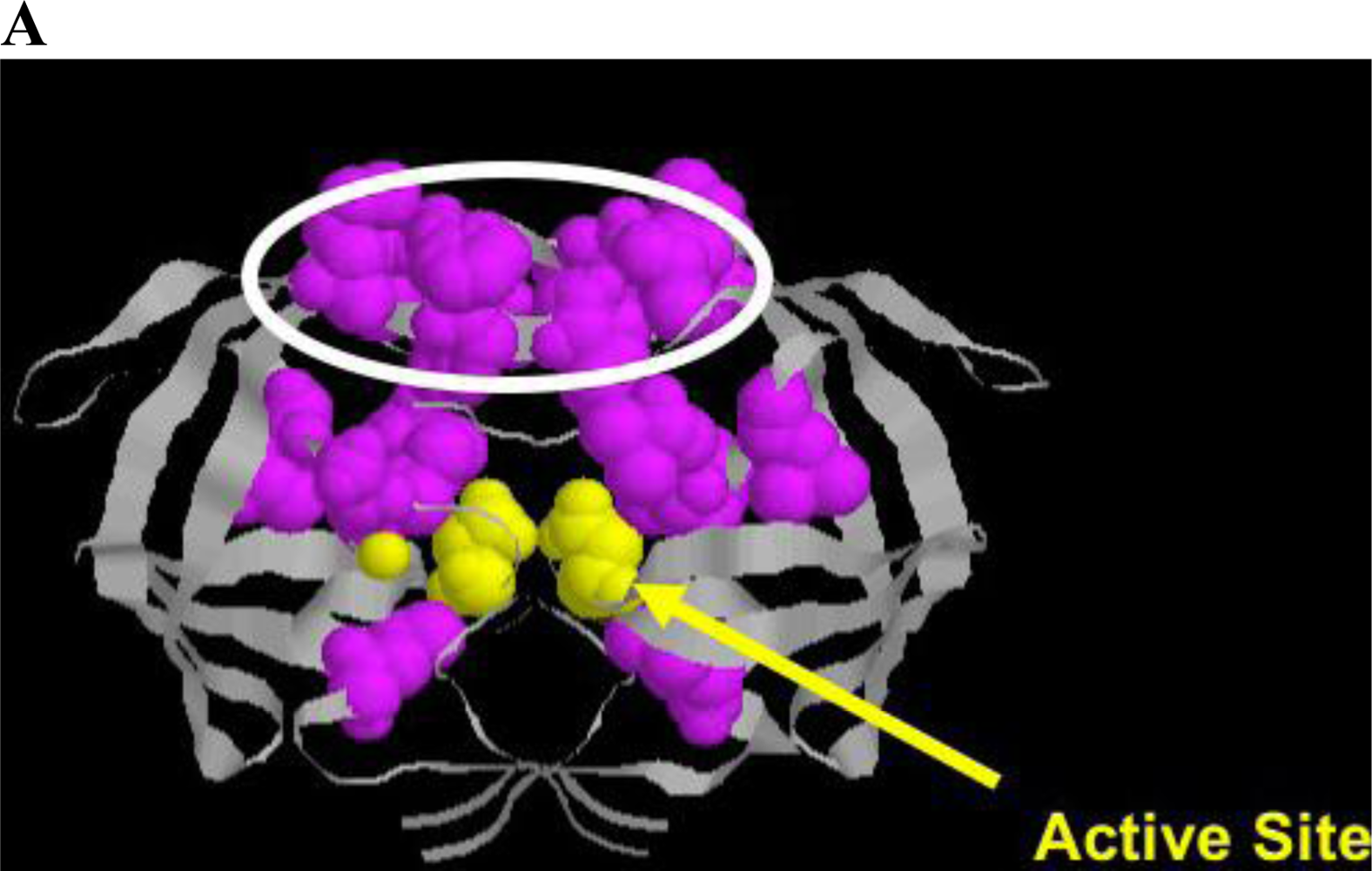
4.3. Protease
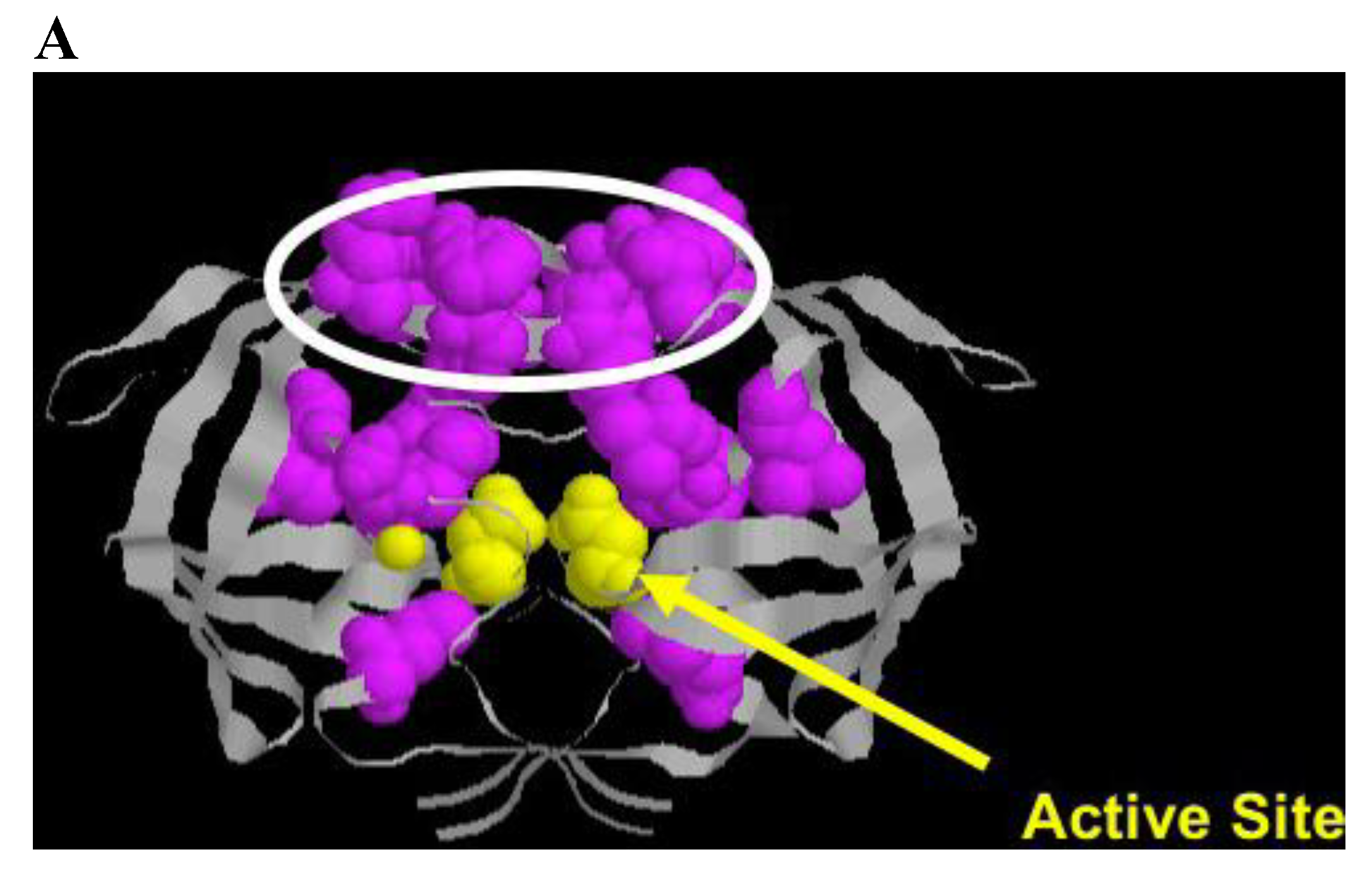
HIV protease processes the Gag and Gag/Pol protein precursors, resulting in virus maturation and virus infectivity. Protease inhibitors, either peptidometic or nonpeptidometic, are designed to bind to the active site of the protease. Protease is only 99 amino acids in length, but tolerates a relatively large number of mutations that confer resistance (Figure 4A). A substrate fit model developed by Schiffer and colleagues has developed a useful model explaining drug resistance mutations that emerge near the active site and in flap domains of the molecule that provide access to the active site [108,109,110]. In general, mutations conferring resistance (Figure 4B) are divided into primary mutations, generally drug specific, and secondary mutations, which by themselves confer little resistance to therapy, but in the presence of primary mutations result in increased cross resistance to a number of protease inhibitors [111,112]. Thus, switching protease inhibitors carries a measure of phenotypic “baggage” that can affect subsequent protease inhibitor therapy. Additional mutations occur at protease cleavage sites, resulting in increased efficiency of cleavage despite accumulation of changes in the enzyme proper. Ritonavir is a potent inhibitor of the cyp3A4 metabolic pathway metabolizing many protease inhibitors, and the use of ritonavir to pharmacologically boost protease inhibitor levels in patients has been a useful strategy to maintain protease inhibitor levels and permit once daily dosing. New agents with similar activity are under consideration, including one in clinical trials [113].
4.4. Coreceptor Inhibitors
Maraviroc inhibits binding of HIV gp120 to the CCR5 coreceptor of HIV. As HIV variants may utilize either CCR5 or CXCR4 (or both, “dualtropic” viruses) as coreceptor, resistance to Maraviroc may occur as a population shift to CXCR4-tropic or dual tropic viruses. Sensitive tropism assays are available to determine presence of CXCR4 or dual tropic viruses at relatively low frequencies (reported at 0.1%) [115]. Such tropism assays are required prior to initiating therapy, as the presence of dual tropic or CXCR4 tropic virus compromises Maraviroc efficacy. In addition, resistance can occur by emergence of mutations that affect binding of Maraviroc. Thus, the etiology of rebound viremia on Maraviroc or other coreceptor inhibitors may be multifactorial.
4.5. Fusion Inhibitors
Enfuvirtide [116] is a 36 amino acid peptide that binds to alpha helix within gp41, disrupting the “spring-loaded” like mechanism that mediates viral-cell fusion [116,117]. Early studies identified mutations emerging in the binding domain abrogating enfuvirtide activity, and found such changes in great majority of patients with rebound viremia while adherent on enfuvirtide therapy [118,119]. Thus it is typically not cost effective to genotype rebound viremia for T-20 resistance mutations. Deeks and coworkers demonstrated continued T-20 antiviral activity even in the presence of mutations [120]. It is likely that drug resistance mutations reduce replication capacity; the potential clinical benefit of maintaining T-20 despite resistance is weighed against the inconvenience, discomfort, and relative difficulty in twice daily subcutaneous administration.
4.6. Integrase Inhibitors
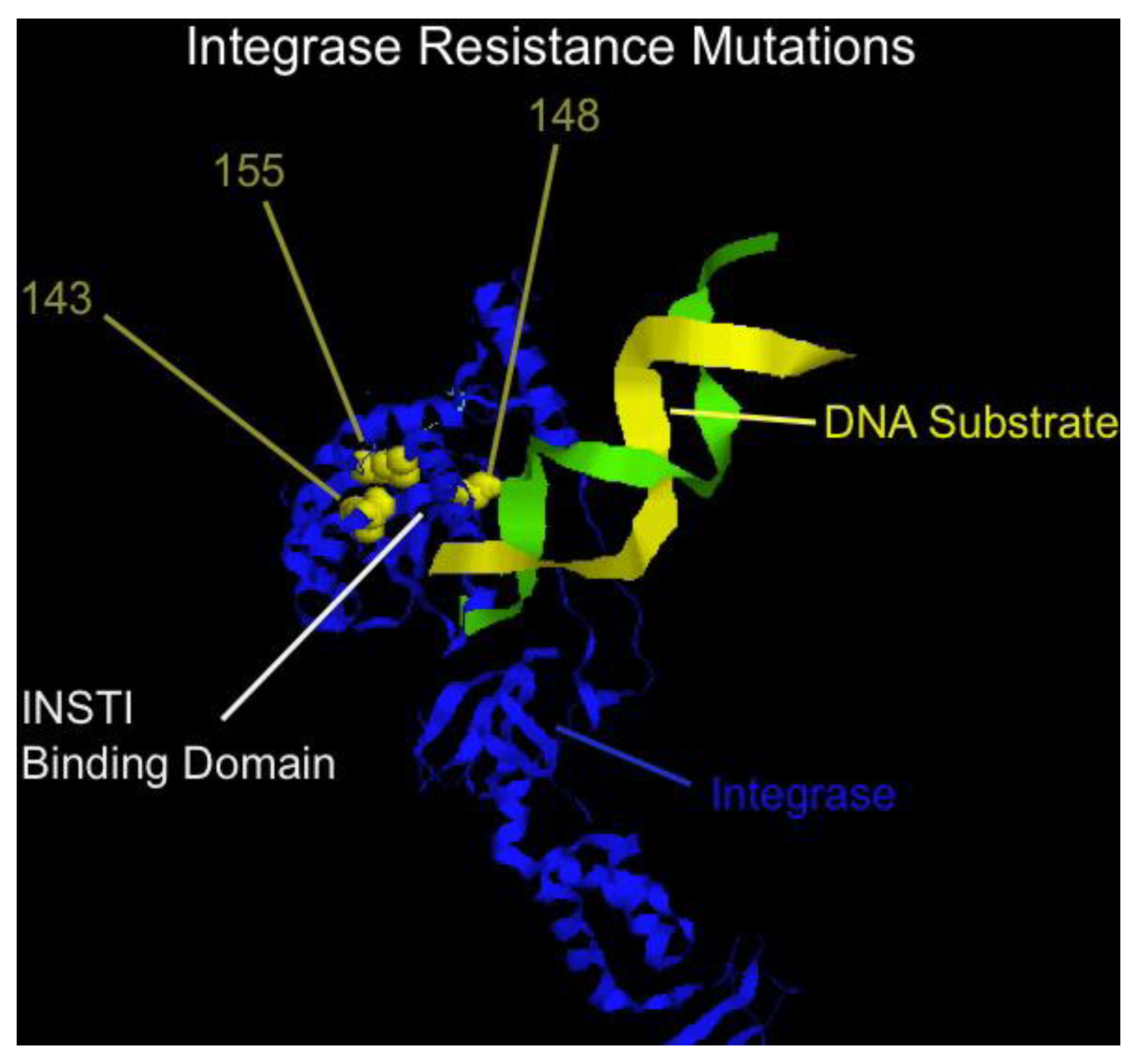
Raltegravir is the first in class integrase strand transfer inhibitors (INSTI) and represents a highly potent and well tolerated addition to the antiviral armamentarium [121,122,123]. A second INSTI, elvitegravir, is currently in late stage development [124]. Despite the marked activity and ease of tolerability, raltegravir is, however, similar to all antiretrovirals in that resistance can emerge relatively quickly if the drug is used in combinations with ineffective or recycled antivirals. A number of independent pathways to resistance have been identified, with mutations at positions N155H, Q148H/R/K, or Y143R/H/C yielding high level resistance to therapy, typically with a number of additional secondary mutations [125]. Mutations map near the binding site of the inhibitor (Figure 5), and a common binding site for raltegravir and elvitegravir explains, in part, the cross-resistance between these two inhibitors [126]. Additional INSTI are in development, at least one of which has reported to have some evidence of non-cross resistance to raltegravir [127]. Phenotyping for INSTI are now commercially available.
5. Non-Subtype B and HIV-2 Infections
5.1. Non-Subtype B Infection
Although the vast majority of genotypic information has been obtained using HIV group M, subtype B, investigations of non-B viruses are ongoing and increasing [129,130]. A number of differences in drug resistance have been reported [131]; for example the rate in which certain mutations occur, e.g., K65R appears to occur in subtype C faster than in subtype B [130], and the NNRTI drug resistance mutation V106M is commonly identified in subtype C but not B [132,133]; the emergence of either mutation may be explained, in part by the baseline wild- type sequence favoring the development of the mutation. In general, however, non-B subtypes have similar sensitivities and responses to antiretrovirals as subtype B [134,135,136,137,138]. Non-M viruses have not been extensively studied, although group O viruses are intrinsically resistant to the NNRTI nevirapine and efavirenz. Initial data on etravirine [139] and raltegravir [140] resistance in non-B viruses has been reported.
5.2. HIV-2 Infection
HIV-2 infection shows striking differences in drug sensitivity from HIV-1. Although some variability among isolates exists, HIV-2 is intrinsically resistant to NNRTIs; susceptibility to etravirine has not been studied extensively. NRTI resistance emerges in HIV-2, and has a higher prevalence of multidrug resistance Q151M suite of mutations (A62V, V75I, F77L, F116Y, Q151M) [141,142].
6. Clinical Management of Drug Resistance
Clinical management of HIV drug resistance proceeds as with other clinical diseases, and begins with a comprehensive history, followed by physical examination and indicated laboratory studies. In general, patients undergoing successful combination antiretroviral therapy have viral RNA levels suppressed below the commercial limit of detection (50–75 copies HIV-1 RNA/mL plasma) using either bDNA or RT-PCR based methodologies.
The first indication that drug resistance may be present is an increase in plasma viremia above the clinical cutoff during a routine clinical visit. The turnaround time for HIV RNA levels determination is at least several days, and results from testing will not be available if the clinical visit is timed with the phlebotomy for HIV RNA. One approach to this delay from phlebotomy to result is to obtain blood for viral RNA levels, and CD4 cell numbers one week prior to the clinical visit, at which time the clinical evaluation with all relevant laboratories studies may be obtained. In this way, the patient with rebound viremia may be counseled in person from the outset.
All increases in viremia should be evaluated as soon as possible, but not all increases in viremia represent drug resistance and other causes are typically ruled out prior to in depth evaluation for resistance, and repeat viral RNA testing is essential. Low level increases in viremia (blips, viral RNA 50-ca. 200 copies HIV RNA/mL plasma) may be the result of assay variation, and do not often reflect true rebound viremia [143]. Recent development of a new version of the Abbott amplicor PCR increases in viral RNA have been noted in the 74–200 copy range [144,145,146]. These increases have not been sustained or associated with resistance [147] and may reflect statistical variation near the limit of detection. Intercurrent, typically febrile, illnesses represent a second cause of rebound viremia. The precise cause of increased viremia remains uncertain, as not all illnesses result in elevations in viremia; rebounds that do occur are not associated with drug resistance, and may be the result of generalized immune activation. A third cause of rebound viremia not due to drug resistance is antiretroviral nonadherence. A thorough history and physical examination, to rule out antecedent illness or nonadherence coupled with repeat viral RNA testing represents the initial evaluation of rebound viremia.
Sustained increases in HIV viremia in the absence of other potential causes suggests that resistance mutations conferring drug resistance have emerged. Low level rebound viremia typically continues to increase, which prompts resistance testing. A minimum of viremia is essential for resistance testing; there are no absolute limits although levels ca. 500 copies/mL may be unreliably amplified, and tropism testing requires viremia in excess of 1000 copies/mL plasma. It is difficult to predict which component of drug regimen resistance may have failed during rebound viremia. In general, resistance mutations that confer high level resistance (e.g., K103N, efavirenz or M184V, 3TC/FTC) are likely to be present during rebound viremia. Resistance to other antiretrovirals is more difficult to predict. In addition, as described above, understanding of specific drug resistance mutations that emerge on therapy to specific agents may provide some virologic benefit in the setting of other agents. For instance, development of M184V during 3TC/FTC therapy results in a less fit virus, and one which, in the presence of TAMS, is more sensitive to NRTI therapy with AZT, D4T, or tenofovir. Similarly, K65R-containing virus arising from from tenofovir, abacavir or ddI is more sensitive to AZT than the wild type virus [45,148], T215Y/F-containing virus from AZT therapy is more sensitive to tenofovir than wild type virus. As a consequence, 3TC or FTC is often continued in individuals with M184V because of the potential antiviral effect. Tenofovir and AZT have been used in treatment experienced individuals to suppress T215Y/F and K65R. The results of these laboratory tests represent useful data, but are not themselves designed to be clinical management tools. Choice of therapy for treatment experienced individuals represents a cooperative interaction between patients and health care professionals and requires assessment of resistance, drug tolerability, adverse effect profile of potential agents, and drug interactions. Regimens are often constructed around development of newly developed agents directed against novel viral targets, but without additional active agents, the new regimen is effectively monotherapy; viral suppression is typically transient and efficacy from the new drug may be completely lost. In general, two new agents to which the patient does not have resistance are used for therapy for experienced patients [85]. With extensive cross resistance among drug classes, the goal of reducing viremia to <50 copies/mL plasma with two fully active agents, is often not possible. Despite adverse effects, partially active therapy is clearly superior to drug interruptions; useful guidelines are available to assist antiretroviral choice [85,86].
As described, therapy for HIV infection is individualized based on firm basic and clinical science. Guidelines for the use of antiretroviral therapy have been developed and are regularly updated; panels of experts review and debate accumulated evidence and provide recommendations regarding use of antiretrovirals and resistance testing. Several commendable aspects of this process include: Graded recommendations, e.g., critical recommendations well founded by randomized controlled trials are weighted with high confidence. Recommendations that are based on “expert opinion” are marked with lower confidence. In addition, some guidelines include a highlighted discussion of new or changed recommendations, providing knowledgeable practitioners with a rapid mechanism to update their fund of knowledge. The US Department of Health and Human Services guidelines is regularly updated in a “living-document” fashion [149]. As a consequence, providers worldwide have access to current and useful information.
7. Unresolved Issues
Although several clear instances of pre-existing mutations contributes to emergence of drug resistance, not all resistance is clearly explained by pre-existing resistance mutations, and the role of low level drug resistance mutations remains undefined. Genetic composition of HIV from drug resistant individuals is complex but can be investigated with sensitive techniques [13,14,150,151,152,153]. In one sense, since most of single drug resistance mutations are likely present in large genetically diverse virus populations [10,154], the real question is what level of resistance is associated with clinical resistance [155]. Cross-sectional studies of low level transmitted drug resistance was associated with regimen failure [156,157], and recent analysis of ACTG5095 [158] demonstrated increased risk of regimen failure in the presence of low level NNRTI mutations, and analysis of ACTG5208, randomized study of combination therapy for individuals with prior nevirapine exposure revealed relatively low level resistance [159] was associated with risk of viral rebound or death [160]. Application of new techniques, such as massively parallel pyrosequencing, may shed new light on the role of minority resistance or tropism variants in emergence of HIV drug resistance [161,162,163,164].
Viral rebound or viral suppression may occur [165] in the presence of a partially suppressive regimen. For instance, patients taking NNRTI based regimens may experience viral rebound; analysis of drug resistance may reveal emergence of a single nucleotide change conferring drug resistance [11]. Continued suppression; however, may also occur in the presence of a single mutation such as K103N. Why viremia rebounds in some patients but not others with known mutations is unknown.
The relative contributions of individual antiretroviral during rebound viremia remains uncertain, but such data might be most useful in constructing new regimens. Whether some drugs (e.g., tenofovir, protease inhibitors, INSTI) continue to have efficacy in the setting of accumulated resistance mutations has not been well described. Contributions of HIV fitness to viral replication and pathogenesis, reviewed in [166] are clear. Sensitive viral fitness assays are useful in understanding the effects of mutations on viral replication and pathogenesis [167,168,169,170], and in understanding replication of viruses from individuals [171] but are not feasible for routine clinical use. Relative replication capacity measurements are made in some phenotyping assays. When fully suppressive regimens are not possible, choosing regimens yielding lowest replication capacity may be desirable; in early studies, higher CD4 cell numbers were associated with lower replication capacity [172,173], and accumulation of drug resistance mutations is associated with lower replication capacity [125,173,174] but clear evidence for clinical benefit of lower relative replication capacity remains under study.
New bioinformatics approaches to analyze genotypic and phenotypic data continue to be investigated [72]. In general, resistance testing analyzes individual drugs singly, not in combination. New artificial neural networks are in development that incorporate resistance data with clinical information to identify entire regimens in a patient specific manner [175,176,177]. The use of induction-maintenance therapeutic strategies has resulted in persistent suppression for prolonged periods [159,178,179], but how a single agent maintains suppression in a substantial proportion of patients remains uncertain. Continued studies of population dynamics and the potential contribution of immune responses are likely to shed new light on the nature of drug resistance in vivo.
Acknowledgments
We thank our patients and the NIAID Clinical Infectious Disease fellows for stimulating discussions.
References
- Broder, S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antivir. Res. 2010, 85, 1–18. [Google Scholar] [CrossRef]
- Tsibris, A.M.; Hirsch, M.S. Antiretroviral therapy in the clinic. J. Virol. 2010, 84, 5458–5464. [Google Scholar] [CrossRef] [PubMed]
- Volberding, P.A.; Deeks, S.G. Antiretroviral therapy and management of HIV infection. Lancet 2010, 376, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Paredes, R.; Clotet, B. Clinical management of HIV-1 resistance. Antivir. Res. 2010, 85, 245–265. [Google Scholar] [CrossRef] [PubMed]
- Hogg, R.S.; Bangsberg, D.R.; Lima, V.D.; Alexander, C.; Bonner, S.; Yip, B.; Wood, E.; Dong, W.W.; Montaner, J.S.; Harrigan, P.R. Emergence of drug resistance is associated with an increased risk of death among patients first starting HAART. PLoS Med. 2006, 3, e356. [Google Scholar] [CrossRef] [PubMed]
- Little, S.J.; Frost, S.D.; Wong, J.K.; Smith, D.M.; Pond, S.L.; Ignacio, C.C.; Parkin, N.T.; Petropoulos, C.J.; Richman, D.D. Persistence of transmitted drug resistance among subjects with primary human immunodeficiency virus infection. J. Virol. 2008, 82, 5510–5518. [Google Scholar] [CrossRef]
- Vo, T.T.; Ledergerber, B.; Keiser, O.; Hirschel, B.; Furrer, H.; Battegay, M.; Cavassini, M.; Bernasconi, E.; Vernazza, P.; Weber, R. Durability and outcome of initial antiretroviral treatments received during 2000–2005 by patients in the Swiss HIV Cohort Study. J. Infect. Dis. 2008, 197, 1685–1694. [Google Scholar] [CrossRef]
- Huang, H.Y.; Daar, E.S.; Sax, P.E.; Young, B.; Cook, P.; Benson, P.; Cohen, C.; Scribner, A.; Hu, H. The prevalence of transmitted antiretroviral drug resistance in treatment-naive patients and factors influencing first-line treatment regimen selection. HIV Med. 2008, 9, 285–293. [Google Scholar] [CrossRef]
- Perelson, A.S.; Neumann, A.U.; Markowitz, M.; Leonard, J.M.; Ho, D.D. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science 1996, 271, 1582–1586. [Google Scholar] [CrossRef]
- Bonhoeffer, S.; Coffin, J.M.; Nowak, M.A. Human immunodeficiency virus drug therapy and virus load. J. Virol. 1997, 71, 3275–3278. [Google Scholar] [CrossRef]
- Coffin, J.M. HIV viral dynamics. AIDS 1996, 10, S75–S84. [Google Scholar] [PubMed]
- Frost, S.D.; Nijhuis, M.; Schuurman, R.; Boucher, C.A.; Brown, A.J. Evolution of lamivudine resistance in human immunodeficiency virus type 1-infected individuals: The relative roles of drift and selection. J. Virol. 2000, 74, 6262–6268. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Li, J.F.; Morris, L.; Martinson, N.; Gray, G.; McIntyre, J.; Heneine, W. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. J. Infect. Dis. 2005, 192, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Boltz, V.; Maldarelli, F.; Kearney, M.; Halvas, E.K.; Rock, D.; Falloon, J.; Davey, R.T.Jr.; Dewar, R.L.; Metcalf, J.A.; Mellors, J.W.; Coffin, J.M. Selection and persistence of non-nucleoside reverse transcriptase inhibitor-resistant HIV-1 in patients starting and stopping non-nucleoside therapy. AIDS 2006, 20, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Rhodes, T.; Dang, Q.; Pathak, V. Retroviral recombination: Review of genetic analyses. Front. Biosci. 2003, 8, d143–d155. [Google Scholar] [CrossRef]
- Barry, M.; Gibbons, S.; Back, D.; Mulcahy, F. Protease inhibitors in patients with HIV disease. Clinically important pharmacokinetic considerations. Clin. Pharmacokinet. 1997, 32, 194–209. [Google Scholar] [CrossRef]
- Preston, S.L.; Piliero, P.J.; Drusano, G.L. Pharmacodynamics and clinical use of anti-HIV drugs. Infect. Dis. Clin. North Am. 2003, 17, 651–674. [Google Scholar] [CrossRef] [PubMed]
- Demeter, L.M.; Jiang, H.; Mukherjee, A.L.; Morse, G.D.; DiFrancesco, R.; DiCenzo, R.; Dykes, C.; Sista, P.; Bacheler, L.; Klingman, K.; Rinehart, A.; Albrecht, M. A randomized trial of therapeutic drug monitoring of protease inhibitors in antiretroviral-experienced, HIV-1-infected patients. AIDS 2009, 23, 357–368. [Google Scholar] [CrossRef]
- Kredo, T.; Van der Walt, J.S.; Siegfried, N.; Cohen, K. Therapeutic drug monitoring of antiretrovirals for people with HIV. Cochrane Database Syst. Rev. 2009, CD007268. [Google Scholar] [CrossRef]
- Simoni, J.M.; Amico, K.R.; Pearson, C.R.; Malow, R. Strategies for promoting adherence to antiretroviral therapy: a review of the literature. Curr. Infect. Dis. Rep. 2008, 10, 515–521. [Google Scholar] [CrossRef]
- Simoni, J.M.; Amico, K.R.; Smith, L.; Nelson, K. Antiretroviral adherence interventions: Translating research findings to the real world clinic. Curr. HIV/AIDS Rep. 2010, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.E.; Jeon, C.Y.; Ivers, L.C.; Behforouz, H.L.; Caldas, A.; Drobac, P.C.; Shin, S.S. Effect of directly observed therapy for highly active antiretroviral therapy on virologic, immunologic, and adherence outcomes: a meta-analysis and systematic review. J. Acquir. Immune Defic. Syndr. 2010, 54, 167–179. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, M.; Viechtbauer, W.; Schaalma, H.P.; Kok, G.; Abraham, C.; Hospers, H.J. Standard care impact on effects of highly active antiretroviral therapy adherence interventions: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2010, 170, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.; Deeks, S.G.; van der Laan, M.; Bangsberg, D.R. The risk of virologic failure decreases with duration of HIV suppression, at greater than 50% adherence to antiretroviral therapy. PLoS ONE 2009, 4, e7196. [Google Scholar] [CrossRef]
- Bello, G.; Casado, C.; Sandonis, V.; Alvaro-Cifuentes, T.; Dos Santos, C.A.; Garcia, S.; Rodriguez, C.; Del Romero, J.; Pilotto, J.H.; Grinsztejn, B.; Veloso, V.G.; Morgado, M.G.; Lopez-Galindez, C. Plasma viral load threshold for sustaining intrahost HIV type 1 evolution. AIDS Res. Hum. Retroviruses 2007, 23, 1242–1250. [Google Scholar] [CrossRef]
- Persaud, D.; Pierson, T.; Ruff, C.; Finzi, D.; Chadwick, K.R.; Margolick, J.B.; Ruff, A.; Hutton, N.; Ray, S.; Siliciano, R.F. A stable latent reservoir for HIV-1 in resting CD4(+) T lymphocytes in infected children. J. Clin. Invest. 2000, 105, 995–1003. [Google Scholar] [CrossRef]
- Martinez-Picado, J.; DePasquale, M.P.; Kartsonis, N.; Hanna, G.J.; Wong, J.; Finzi, D.; Rosenberg, E.; Gunthard, H.F.; Sutton, L.; Savara, A.; Petropoulos, C.J.; Hellmann, N.; Walker, B.D.; Richman, D.D.; Siliciano, R.; D’Aquila, R.T. Antiretroviral resistance during successful therapy of HIV type 1 infection. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10948–10953. [Google Scholar] [CrossRef]
- Shen, L.; Siliciano, R.F. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J. Allergy Clin. Immunol. 2008, 122, 22–28. [Google Scholar] [CrossRef]
- Maldarelli, F. Targeting viral reservoirs: Ability of antiretroviral therapy to stop viral replication. Curr. Opin. HIV AIDS 2011, 6, 49–56. [Google Scholar] [CrossRef]
- Buzon, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; Palmer, S.; Stevenson, M.; Clotet, B.; Blanco, J.; Martinez-Picado, J. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat. Med. 2010, 16, 460–465. [Google Scholar] [CrossRef]
- Lakhman, S.S.; Ma, Q.; Morse, G.D. Pharmacogenomics of CYP3A: Considerations for HIV treatment. Pharmacogenomics 2009, 10, 1323–1339. [Google Scholar] [CrossRef] [PubMed]
- Aihara, M. Pharmacogenetics of cutaneous adverse drug reactions. J. Dermatol. 2011, 38, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.M.; Kuritzkes, D.R.; Johnson, V.A.; Mellors, J.W.; Sullivan, J.L.; Swanstrom, R.; D’Aquila, R.T.; Van Gorder, M.; Holodniy, M.; Lloyd Jr, R.M.Jr.; Reid, C.; Morgan, G.F.; Winslow, D.L. Accuracy of the TRUGENE HIV-1 genotyping kit. J. Clin. Microbiol. 2003, 41, 1586–1593. [Google Scholar] [CrossRef]
- Pandit, A.; Mackay, W.G.; Steel, C.; van Loon, A.M.; Schuurman, R. HIV-1 drug resistance genotyping quality assessment: results of the ENVA7 Genotyping Proficiency Programme. J. Clin. Virol. 2008, 43, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.A.; Aberg, J.A.; Cahn, P.; Montaner, J.S.; Rizzardini, G.; Telenti, A.; Gatell, J.M.; Gunthard, H.F.; Hammer, S.M.; Hirsch, M.S.; Jacobsen, D.M.; Reiss, P.; Richman, D.D.; Volberding, P.A.; Yeni, P.; Schooley, R.T. Antiretroviral treatment of adult HIV infection: 2010 recommendations of the International AIDS Society-USA panel. JAMA 2010, 304, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Labayru, C.; Eiros, J.M.; Hernandez, B.; de Lejarazu, R.O.; Torres, A.R. RNA extraction prior to HIV-1 resistance detection using Line Probe Assay (LiPA): comparison of three methods. J. Clin. Virol. 2005, 32, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, M.S.; Troyer, R.M.; Syed, A.R.; Bulime, S.; Demers, K.; Bajunirwe, F.; Arts, E.J. Sensitive oligonucleotide ligation assay for low-level detection of nevirapine resistance mutations in human immunodeficiency virus type 1 quasispecies. J. Clin. Microbiol. 2007, 45, 2604–2615. [Google Scholar] [CrossRef] [PubMed]
- Re, M.C.; Monari, P.; Bon, I.; Gibellini, D.; Vitone, F.; Borderi, M.; La Placa, M. Analysis of HIV-1 drug resistant mutations by line probe assay and direct sequencing in a cohort of therapy naive HIV-1 infected Italian patients. BMC Microbiol. 2001, 1, 30. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef]
- Fisher, A.G.; Collalti, E.; Ratner, L.; Gallo, R.C.; Wong-Staal, F. A molecular clone of HTLV-III with biological activity. Nature 1985, 316, 262–265. [Google Scholar] [CrossRef]
- Brun-Vezinet, F.; Costagliola, D.; Khaled, M.A.; Calvez, V.; Clavel, F.; Clotet, B.; Haubrich, R.; Kempf, D.; King, M.; Kuritzkes, D.; Lanier, R.; Miller, M.; Miller, V.; Phillips, A.; Pillay, D.; Schapiro, J.; Scott, J.; Shafer, R.; Zazzi, M.; Zolopa, A.; DeGruttola, V. Clinically validated genotype analysis: guiding principles and statistical concerns. Antivir. Ther. 2004, 9, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, J.; Vandamme, A.M. Algorithms for the interpretation of HIV-1 genotypic drug resistance information. Antivir. Res. 2006, 71, 335–342. [Google Scholar] [CrossRef] [PubMed]
- HIV Drug Resistance Website. Available online: http://hivdb.stanford.edu (accessed on 21 February 2011).
- Garcia-Lerma, J.G.; Nidtha, S.; Blumoff, K.; Weinstock, H.; Heneine, W. Increased ability for selection of zidovudine resistance in a distinct class of wild-type HIV-1 from drug-naive persons. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13907–13912. [Google Scholar] [CrossRef] [PubMed]
- Stephan, C.; Dauer, B.; Bickel, M.; Haberl, A.; Locher, L.; Muller, A.; Klauke, S.; Berger, A.; Doerr, H.W.; Sturmer, M.; Staszewski, S. Intensification of a failing regimen with zidovudine may cause sustained virologic suppression in the presence of resensitising mutations including K65R. J. Infect. 2010, 61, 346–350. [Google Scholar] [CrossRef]
- Shi, C.; Mellors, J.W. A recombinant retroviral system for rapid in vivo analysis of human immunodeficiency virus type 1 susceptibility to reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 1997, 41, 2781–2785. [Google Scholar] [CrossRef]
- Hertogs, K.; de Bethune, M.P.; Miller, V.; Ivens, T.; Schel, P.; Van Cauwenberge, A.; Van Den Eynde, C.; Van Gerwen, V.; Azijn, H.; et al. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicrob. Agents Chemother. 1998, 42, 269–276. [Google Scholar] [CrossRef]
- Petropoulos, C.J.; Parkin, N.T.; Limoli, K.L.; Lie, Y.S.; Wrin, T.; Huang, W.; Tian, H.; Smith, D.; Winslow, G.A.; Capon, D.J.; Whitcomb, J.M. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2000, 44, 920–928. [Google Scholar] [CrossRef]
- Kellam, P.; Larder, B.A. Recombinant virus assay: A rapid, phenotypic assay for assessment of drug susceptibility of human immunodeficiency virus type 1 isolates. Antimicrob. Agents Chemother. 1994, 38, 23–30. [Google Scholar] [CrossRef]
- Debiaggi, M.; Bruno, R.; Sacchi, P.; Achilli, G.; Romero, E.; Filice, G. Distinct mutational drug resistance profiles of HIV-1 RNA in plasma and culture isolates of patients receiving antiretroviral therapy. Intervirology 2002, 45, 52–55. [Google Scholar] [CrossRef]
- Qari, S.H.; Respess, R.; Weinstock, H.; Beltrami, E.M.; Hertogs, K.; Larder, B.A.; Petropoulos, C.J.; Hellmann, N.; Heneine, W. Comparative analysis of two commercial phenotypic assays for drug susceptibility testing of human immunodeficiency virus type 1. J. Clin. Microbiol. 2002, 40, 31–35. [Google Scholar] [CrossRef]
- Robinson, L.H.; Myers, R.E.; Snowden, B.W.; Tisdale, M.; Blair, E.D. HIV type 1 protease cleavage site mutations and viral fitness: implications for drug susceptibility phenotyping assays. AIDS Res. Hum. Retroviruses 2000, 16, 1149–1156. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; King, N.M.; Schiffer, C.A. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 2004, 78, 12446–12454. [Google Scholar] [CrossRef] [PubMed]
- Kolli, M.; Stawiski, E.; Chappey, C.; Schiffer, C.A. Human immunodeficiency virus type 1 protease-correlated cleavage site mutations enhance inhibitor resistance. J. Virol. 2009, 83, 11027–11042. [Google Scholar] [CrossRef] [PubMed]
- Swanstrom, R.; Bosch, R.J.; Katzenstein, D.; Cheng, H.; Jiang, H.; Hellmann, N.; Haubrich, R.; Fiscus, S.A.; Fletcher, C.V.; Acosta, E.P.; Gulick, R.M. Weighted phenotypic susceptibility scores are predictive of the HIV-1 RNA response in protease inhibitor-experienced HIV-1-infected subjects. J. Infect. Dis. 2004, 190, 886–893. [Google Scholar] [CrossRef]
- Harrigan, P.R.; Montaner, J.S.; Wegner, S.A.; Verbiest, W.; Miller, V.; Wood, R.; Larder, B.A. World-wide variation in HIV-1 phenotypic susceptibility in untreated individuals: biologically relevant values for resistance testing. AIDS 2001, 15, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Winters, B.; Montaner, J.; Harrigan, P.R.; Gazzard, B.; Pozniak, A.; Miller, M.D.; Emery, S.; van Leth, F.; Robinson, P.; Baxter, J.D.; et al. Determination of clinically relevant cutoffs for HIV-1 phenotypic resistance estimates through a combined analysis of clinical trial and cohort data. J. Acquir. Immune Defic. Syndr. 2008, 48, 26–34. [Google Scholar] [CrossRef]
- Mazzotta, F.; Lo Caputo, S.; Torti, C.; Tinelli, C.; Pierotti, P.; Castelli, F.; Lazzarin, A.; Angarano, G.; Maserati, R.; Gianotti, N.; et al. Real versus virtual phenotype to guide treatment in heavily pretreated patients: 48-week follow-up of the Genotipo-Fenotipo di Resistenza (GenPheRex) trial. J. Acquir. Immune Defic. Syndr. 2003, 32, 268–280. [Google Scholar] [CrossRef]
- Saracino, A.; Monno, L.; Locaputo, S.; Torti, C.; Scudeller, L.; Ladisa, N.; Antinori, A.; Sighinolfi, L.; Chirianni, A.; Mazzotta, F.; Carosi, G.; Angarano, G. Selection of antiretroviral therapy guided by genotypic or phenotypic resistance testing: an open-label, randomized, multicenter study (PhenGen). J. Acquir. Immune Defic. Syndr. 2004, 37, 1587–1598. [Google Scholar] [CrossRef]
- Torti, C.; Quiros-Roldan, E.; Regazzi, M.; De Luca, A.; Mazzotta, F.; Antinori, A.; Ladisa, N.; Micheli, V.; Orani, A.; Patroni, A.; Villani, P.; Lo Caputo, S.; Moretti, F.; Di Giambenedetto, S.; Castelnuovo, F.; Maggi, P.; Tinelli, C.; Carosi, G. A randomized controlled trial to evaluate antiretroviral salvage therapy guided by rules-based or phenotype-driven HIV-1 genotypic drug-resistance interpretation with or without concentration-controlled intervention: the Resistance and Dosage Adapted Regimens (RADAR) study. Clin. Infect. Dis. 2005, 40, 1828–1836. [Google Scholar]
- Beerenwinkel, N.; Daumer, M.; Oette, M.; Korn, K.; Hoffmann, D.; Kaiser, R.; Lengauer, T.; Selbig, J.; Walter, H. Geno2pheno: Estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic. Acids Res. 2003, 31, 3850–3855. [Google Scholar] [CrossRef]
- geno2pheno 3.2. Available online: www.geno2pheno.org (accessed on 21 February, 2011).
- HIV French Resistance Website. Available online: http://www.hivfrenchresistance.org (accessed on 21 February, 2011).
- REGA. Available online: http://regaweb.med.kuleuven.be/software/rega_algorithm/ (accessed on 21 February 2011).
- Altmann, A.; Sing, T.; Vermeiren, H.; Winters, B.; Van Craenenbroeck, E.; Van der Borght, K.; Rhee, S.Y.; Shafer, R.W.; Schulter, E.; Kaiser, R.; et al. Advantages of predicted phenotypes and statistical learning models in inferring virological response to antiretroviral therapy from HIV genotype. Antivir. Ther. 2009, 14, 273–283. [Google Scholar] [CrossRef]
- Liu, T.F.; Shafer, R.W. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- IASUSA. Available online: http://www.iasusa.org/resistance_mutations/index.html (accessed on 21 February, 2011).
- Maldarelli, F. HIV drug resistance. In Handbook of Pediatric HIV Care; Zeichner, S., Read, J., Eds.; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Hales, G.; Birch, C.; Crowe, S.; Workman, C.; Hoy, J.F.; Law, M.G.; Kelleher, A.D.; Lincoln, D.; Emery, S. A randomised trial comparing genotypic and virtual phenotypic interpretation of HIV drug resistance: the CREST study. PLoS Clin. Trials 2006, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.; Isaacson, J.; Brun, S.; Bernstein, B.; Lam, W.; Bertz, R.; Foit, C.; Rynkiewicz, K.; Richards, B.; King, M.; Rode, R.; Kempf, D.J.; Granneman, G.R.; Sun, E. Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 2003, 47, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, A.G.; Cohen-Codar, I.; King, M.S.; Colson, P.; Guillevic, E.; Descamps, D.; Lamotte, C.; Schneider, V.; Ritter, J.; Segondy, M.; et al. Virological and pharmacological parameters predicting the response to lopinavir-ritonavir in heavily protease inhibitor-experienced patients. Antimicrob. Agents Chemother. 2005, 49, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Peterson, S.; Sedaghat, A.R.; McMahon, M.A.; Callender, M.; Zhang, H.; Zhou, Y.; Pitt, E.; Anderson, K.S.; Acosta, E.P.; Siliciano, R.F. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 2008, 14, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Ribaudo, H.J.; Kuritzkes, D.R. Instantaneous inhibitory potential is similar to inhibitory quotient at predicting HIV-1 response to antiretroviral therapy. Clin. Infect. Dis. 2010, 51, 93–98. [Google Scholar] [CrossRef]
- Durant, J.; Clevenbergh, P.; Halfon, P.; Delgiudice, P.; Porsin, S.; Simonet, P.; Montagne, N.; Boucher, C.A.; Schapiro, J.M.; Dellamonica, P. Drug-resistance genotyping in HIV-1 therapy: The VIRADAPT randomised controlled trial. Lancet 1999, 353, 2195–2199. [Google Scholar] [CrossRef]
- Cohen, C.J.; Hunt, S.; Sension, M.; Farthing, C.; Conant, M.; Jacobson, S.; Nadler, J.; Verbiest, W.; Hertogs, K.; Ames, M.; Rinehart, A.R.; Graham, N.M. A randomized trial assessing the impact of phenotypic resistance testing on antiretroviral therapy. AIDS 2002, 16, 579–588. [Google Scholar] [CrossRef]
- Cingolani, A.; Antinori, A.; Rizzo, M.G.; Murri, R.; Ammassari, A.; Baldini, F.; Di Giambenedetto, S.; Cauda, R.; De Luca, A. Usefulness of monitoring HIV drug resistance and adherence in individuals failing highly active antiretroviral therapy: A randomized study (ARGENTA). AIDS 2002, 16, 369–379. [Google Scholar] [CrossRef]
- Meynard, J.L.; Vray, M.; Morand-Joubert, L.; Race, E.; Descamps, D.; Peytavin, G.; Matheron, S.; Lamotte, C.; Guiramand, S.; Costagliola, D.; Brun-Vezinet, F.; Clavel, F.; Girard, P.M. Phenotypic or genotypic resistance testing for choosing antiretroviral therapy after treatment failure: a randomized trial. AIDS 2002, 16, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Tural, C.; Ruiz, L.; Holtzer, C.; Schapiro, J.; Viciana, P.; Gonzalez, J.; Domingo, P.; Boucher, C.; Rey-Joly, C.; Clotet, B. Clinical utility of HIV-1 genotyping and expert advice: the Havana trial. AIDS 2002, 16, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.; Clevenbergh, P.; Garraffo, R.; Halfon, P.; Icard, S.; Del Giudice, P.; Montagne, N.; Schapiro, J.M.; Dellamonica, P. Importance of protease inhibitor plasma levels in HIV-infected patients treated with genotypic-guided therapy: Pharmacological data from the Viradapt Study. AIDS 2000, 14, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.D.; Mayers, D.L.; Wentworth, D.N.; Neaton, J.D.; Hoover, M.L.; Winters, M.A.; Mannheimer, S.B.; Thompson, M.A.; Abrams, D.I.; Brizz, B.J.; Ioannidis, J.P.; Merigan, T.C. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. AIDS 2000, 14, F83–F93. [Google Scholar] [CrossRef]
- Zolopa, A.R.; Lazzeroni, L.C.; Rinehart, A.; Vezinet, F.B.; Clavel, F.; Collier, A.; Conway, B.; Gulick, R.M.; Holodniy, M.; Perno, C.F.; Shafer, R.W.; Richman, D.D.; Wainberg, M.A.; Kuritzkes, D.R. Accuracy, precision, and consistency of expert HIV type 1 genotype interpretation: an international comparison (The GUESS Study). Clin. Infect. Dis. 2005, 41, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Haubrich, R.H.; Kemper, C.A.; Hellmann, N.S.; Keiser, P.H.; Witt, M.D.; Tilles, J.G.; Forthal, D.N.; Leedom, J.; Leibowitz, M.; McCutchan, J.A.; Richman, D.D. A randomized, prospective study of phenotype susceptibility testing versus standard of care to manage antiretroviral therapy: CCTG 575. AIDS 2005, 19, 295–302. [Google Scholar]
- Panidou, E.T.; Trikalinos, T.A.; Ioannidis, J.P. Limited benefit of antiretroviral resistance testing in treatment-experienced patients: a meta-analysis. AIDS 2004, 18, 2153–2161. [Google Scholar] [CrossRef]
- Palella, F.J.Jr.; Armon, C.; Buchacz, K.; Cole, S.R.; Chmiel, J.S.; Novak, R.M.; Wood, K.; Moorman, A.C.; Brooks, J.T. The association of HIV susceptibility testing with survival among HIV-infected patients receiving antiretroviral therapy: A cohort study. Ann. Intern. Med. 2009, 151, 73–84. [Google Scholar] [CrossRef]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. 1 December 2009; 1–161.
- Gazzard, B.G.; Anderson, J.; Babiker, A.; Boffito, M.; Brook, G.; Brough, G.; Churchill, D.; Cromarty, B.; Das, S.; Fisher, M.; et al. British HIV Association Guidelines for the treatment of HIV-1-infected adults with antiretroviral therapy 2008. HIV Med. 2008, 9, 563–608. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Das, K.; Clark, A.D.Jr.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 10027–10032. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M. Mechanisms of resistance associated with excision of incorporated nucleotide analogue inhibitors of HIV-1 reverse transcriptase. Curr. Opin. HIV AIDS 2007, 2, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Theys, K.; Deforche, K.; Libin, P.; Camacho, R.J.; Van Laethem, K.; Vandamme, A.M. Resistance pathways of human immunodeficiency virus type 1 against the combination of zidovudine and lamivudine. J. Gen. Virol. 2010, 91, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Brenner, B.G.; Oliveira, M.; Wainberg, M.A. Lamivudine can exert a modest antiviral effect against human immunodeficiency virus type 1 containing the M184V mutation. Antimicrob. Agents Chemother. 2003, 47, 747–754. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 2008, 134, 124–146. [Google Scholar] [CrossRef]
- Menendez-Arias, L.; Matamoros, T.; Cases-Gonzalez, C.E. Insertions and deletions in HIV-1 reverse transcriptase: consequences for drug resistance and viral fitness. Curr. Pharm. Des. 2006, 12, 1811–1825. [Google Scholar] [CrossRef]
- Miller, M.D.; Margot, N.A.; Hertogs, K.; Larder, B.; Miller, V. Antiviral activity of tenofovir (PMPA) against nucleoside-resistant clinical HIV samples. Nucleos. Nucleot. Nucleic Acids 2001, 20, 1025–1028. [Google Scholar] [CrossRef]
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Palmer, S.; Maldarelli, F.; Fivash, M.J.Jr.; Coffin, J.M.; Pathak, V.K. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3'-azido-3'-deoxythymidine resistance. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 317–322. [Google Scholar] [CrossRef]
- Brehm, J.H.; Koontz, D.; Meteer, J.D.; Pathak, V.; Sluis-Cremer, N.; Mellors, J.W. Selection of mutations in the connection and RNase H domains of human immunodeficiency virus type 1 reverse transcriptase that increase resistance to 3'-azido-3'-dideoxythymidine. J. Virol. 2007, 81, 7852–7859. [Google Scholar] [CrossRef]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Barr, R.; Pathak, V.K. Mutations in human immunodeficiency virus type 1 RNase H primer grip enhance 3'-azido-3'-deoxythymidine resistance. J. Virol. 2007, 81, 6837–6845. [Google Scholar] [CrossRef]
- Ren, J.; Stammers, D.K. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 2008, 134, 157–170. [Google Scholar] [CrossRef]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark, A.D., Jr.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.J. Recent changes to RasMol, recombining the variants. Trends Biochem. Sci. 2000, 25, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Sayle, R.A.; Milner-White, E.J. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 1995, 20, 374. [Google Scholar] [CrossRef]
- Delgado, J.; Shulman, N. NNRTI hypersusceptibility. AIDS Read. 2005, 15, 28–30, 32–24. [Google Scholar] [PubMed]
- Haubrich, R.H.; Jiang, H.; Swanstrom, R.; Bates, M.; Katzenstein, D.; Petch, L.; Fletcher, C.V.; Fiscus, S.A.; Gulick, R.M. Non-nucleoside phenotypic hypersusceptibility cut-point determination from ACTG 359. HIV Clin. Trials 2007, 8, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Shulman, N.S.; Delgado, J.; Bosch, R.J.; Winters, M.A.; Johnston, E.; Shafer, R.W.; Katzenstein, D.A.; Merigan, T.C. Nonnucleoside reverse transcriptase inhibitor phenotypic hypersusceptibility can be demonstrated in different assays. J. Acquir. Immune Defic. Syndr. 2005, 39, 78–81. [Google Scholar] [CrossRef]
- Ambrose, Z.; Herman, B.D.; Sheen, C.W.; Zelina, S.; Moore, K.L.; Tachedjian, G.; Nissley, D.V.; Sluis-Cremer, N. The human immunodeficiency virus type 1 nonnucleoside reverse transcriptase inhibitor resistance mutation I132M confers hypersensitivity to nucleoside analogs. J. Virol. 2009, 83, 3826–3833. [Google Scholar] [CrossRef]
- Clark, S.A.; Shulman, N.S.; Bosch, R.J.; Mellors, J.W. Reverse transcriptase mutations 118I, 208Y, and 215Y cause HIV-1 hypersusceptibility to non-nucleoside reverse transcriptase inhibitors. AIDS 2006, 20, 981–984. [Google Scholar] [CrossRef]
- Demeter, L.M.; DeGruttola, V.; Lustgarten, S.; Bettendorf, D.; Fischl, M.; Eshleman, S.; Spreen, W.; Nguyen, B.Y.; Koval, C.E.; Eron, J.J.; Hammer, S.; Squires, K. Association of efavirenz hypersusceptibility with virologic response in ACTG 368, a randomized trial of abacavir (ABC) in combination with efavirenz (EFV) and indinavir (IDV) in HIV-infected subjects with prior nucleoside analog experience. HIV Clin. Trials 2008, 9, 11–25. [Google Scholar] [CrossRef]
- Anderson, J.; Schiffer, C.; Lee, S.K.; Swanstrom, R. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009, 85–110. [Google Scholar]
- Lefebvre, E.; Schiffer, C.A. Resilience to resistance of HIV-1 protease inhibitors: Profile of darunavir. AIDS Rev. 2008, 10, 131–142. [Google Scholar] [PubMed]
- Nalam, M.N.; Schiffer, C.A. New approaches to HIV protease inhibitor drug design II: testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr. Opin. HIV AIDS 2008, 3, 642–646. [Google Scholar] [CrossRef]
- Shafer, R.W.; Schapiro, J.M. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 2008, 10, 67–84. [Google Scholar] [PubMed]
- Turner, D.; Schapiro, J.M.; Brenner, B.G.; Wainberg, M.A. The influence of protease inhibitor resistance profiles on selection of HIV therapy in treatment-naive patients. Antivir. Ther. 2004, 9, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.A.; German, P.; Murray, B.P.; Wei, L.; Jain, A.; West, S.; Warren, D.; Hui, J.; Kearney, B.P. Pharmacokinetics and pharmacodynamics of GS-9350: A novel pharmacokinetic enhancer without anti-HIV activity. Clin. Pharmacol. Ther. 2010, 87, 322–329. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- Whitcomb, J.M.; Huang, W.; Fransen, S.; Limoli, K.; Toma, J.; Wrin, T.; Chappey, C.; Kiss, L.D.; Paxinos, E.E.; Petropoulos, C.J. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob. Agents Chemother. 2007, 51, 566–575. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Henry, K.; O'Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; Chung, J.; DeMasi, R.; Donatacci, L.; Drobnes, C.; Delehanty, J.; Salgo, M. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef]
- Lazzarin, A.; Clotet, B.; Cooper, D.; Reynes, J.; Arasteh, K.; Nelson, M.; Katlama, C.; Stellbrink, H.J.; Delfraissy, J.F.; Lange, J.; Huson, L.; DeMasi, R.; Wat, C.; Delehanty, J.; Drobnes, C.; Salgo, M. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N. Engl. J. Med. 2003, 348, 2186–2195. [Google Scholar] [CrossRef]
- Melby, T.; Sista, P.; DeMasi, R.; Kirkland, T.; Roberts, N.; Salgo, M.; Heilek-Snyder, G.; Cammack, N.; Matthews, T.J.; Greenberg, M.L. Characterization of envelope glycoprotein gp41 genotype and phenotypic susceptibility to enfuvirtide at baseline and on treatment in the phase III clinical trials TORO-1 and TORO-2. AIDS Res. Hum. Retroviruses 2006, 22, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Rodes, B.; Labernardiere, J.L.; Benito, J.M.; Toro, C.; Gonzalez-Lahoz, J.; Faudon, J.L.; Clavel, F.; Schapiro, J.; Soriano, V. Evolution of genotypic and phenotypic resistance to Enfuvirtide in HIV-infected patients experiencing prolonged virologic failure. J. Med. Virol. 2004, 74, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Lu, J.; Hoh, R.; Neilands, T.B.; Beatty, G.; Huang, W.; Liegler, T.; Hunt, P.; Martin, J.N.; Kuritzkes, D.R. Interruption of enfuvirtide in HIV-1 infected adults with incomplete viral suppression on an enfuvirtide-based regimen. J. Infect. Dis. 2007, 195, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Young, S.D.; Guare, J.P.; Anthony, N.J.; Gomez, R.P.; Wai, J.S.; Vacca, J.P.; Handt, L.; Motzel, S.L.; Klein, H.J.; et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science 2004, 305, 528–532. [Google Scholar] [CrossRef]
- Steigbigel, R.T.; Cooper, D.A.; Kumar, P.N.; Eron, J.E.; Schechter, M.; Markowitz, M.; Loutfy, M.R.; Lennox, J.L.; Gatell, J.M.; Rockstroh, J.K.; et al. Raltegravir with optimized background therapy for resistant HIV-1 infection. N. Engl. J. Med. 2008, 359, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Zahm, J.A.; Bera, S.; Pandey, K.K.; Vora, A.; Stillmock, K.; Hazuda, D.; Grandgenett, D.P. Mechanisms of human immunodeficiency virus type 1 concerted integration related to strand transfer inhibition and drug resistance. Antimicrob. Agents Chemother. 2008, 52, 3358–3368. [Google Scholar] [CrossRef]
- Grant, P.; Zolopa, A. Integrase inhibitors: A clinical review of raltegravir and elvitegravir. J. HIV Ther. 2008, 13, 36–39. [Google Scholar]
- Hatano, H.; Lampiris, H.; Fransen, S.; Gupta, S.; Huang, W.; Hoh, R.; Martin, J.N.; Lalezari, J.; Bangsberg, D.; Petropoulos, C.; Deeks, S.G. Evolution of integrase resistance during failure of integrase inhibitor-based antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2010, 54, 389–393. [Google Scholar] [CrossRef]
- Schafer, J.J.; Squires, K.E. Integrase inhibitors: A novel class of antiretroviral agents. Ann. Pharmacother. 2010, 44, 145–156. [Google Scholar] [CrossRef]
- Clotet, B.; C. K.; Lalezari, J.; Young, B.; Huang, J.; Underwood, M.R.; Ait-Khaled, M.; Nichols, W.G. HIV integrase resistance profiles and S/GSK1349572 baseline phenotypic susceptibility for individuals experiencing virological failure on raltegravir and enrolling in the VIKING phase IIb Pilot study (ING112961). Antivir. Ther. 2010, 15, A:61. [Google Scholar]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.G. Resistance and viral subtypes: How important are the differences and why do they occur? Curr. Opin. HIV AIDS 2007, 2, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cajas, J.L.; Pant-Pai, N.; Klein, M.B.; Wainberg, M.A. Role of genetic diversity amongst HIV-1 non-B subtypes in drug resistance: A systematic review of virologic and biochemical evidence. AIDS Rev. 2008, 10, 212–223. [Google Scholar] [PubMed]
- Bandaranayake, R.M.; Kolli, M.; King, N.M.; Nalivaika, E.; Heroux, A.; Kakizawa, J.; Sugiura, W.; Schiffer, C.A. The Effect of Clade Specific Sequence Polymorphisms on HIV-1 Protease Activity and Inhibitor Resistance Pathways. J. Virol. 2010, 84, 9995–10003. [Google Scholar] [CrossRef]
- Brenner, B.; Turner, D.; Oliveira, M.; Moisi, D.; Detorio, M.; Carobene, M.; Marlink, R.G.; Schapiro, J.; Roger, M.; Wainberg, M.A. A V106M mutation in HIV-1 clade C viruses exposed to efavirenz confers cross-resistance to non-nucleoside reverse transcriptase inhibitors. AIDS 2003, 17, F1–F5. [Google Scholar] [CrossRef]
- Loemba, H.; Brenner, B.; Parniak, M.A.; Ma'ayan, S.; Spira, B.; Moisi, D.; Oliveira, M.; Detorio, M.; Wainberg, M.A. Genetic divergence of human immunodeficiency virus type 1 Ethiopian clade C reverse transcriptase (RT) and rapid development of resistance against nonnucleoside inhibitors of RT. Antimicrob. Agents Chemother. 2002, 46, 2087–2094. [Google Scholar] [CrossRef]
- Champenois, K.; Bocket, L.; Deuffic-Burban, S.; Cotte, L.; Andre, P.; Choisy, P.; Yazdanpanah, Y. Expected response to protease inhibitors of HIV-1 non-B subtype viruses according to resistance algorithms. AIDS 2008, 22, 1087–1089. [Google Scholar] [CrossRef]
- D’Arrigo, R.; Ciccozzi, M.; Gori, C.; Montieri, S.; Aquaro, S.; Bellagamba, R.; Boumis, E.; Di Perri, G.; Pizzi, D.; Antinori, A.; Rezza, G.; Perno, C.F. gp41 sequence variability in HIV type 1 non-B subtypes infected patients undergoing enfuvirtide pressure. AIDS Res. Hum. Retroviruses 2007, 23, 1296–1302. [Google Scholar] [CrossRef]
- Geretti, A.M.; Harrison, L.; Green, H.; Sabin, C.; Hill, T.; Fearnhill, E.; Pillay, D.; Dunn, D. Effect of HIV-1 subtype on virologic and immunologic response to starting highly active antiretroviral therapy. Clin. Infect. Dis. 2009, 48, 1296–1305. [Google Scholar] [CrossRef]
- Holguin, A.; Faudon, J.L.; Labernardiere, J.L.; Soriano, V. Susceptibility of HIV-1 non-B subtypes and recombinant variants to Enfuvirtide. J. Clin. Virol. 2007, 38, 176–180. [Google Scholar] [CrossRef]
- Martinez-Cajas, J.L.; Pai, N.P.; Klein, M.B.; Wainberg, M.A. Differences in resistance mutations among HIV-1 non-subtype B infections: a systematic review of evidence (1996–2008). J. Int. AIDS Soc. 2009, 12, 11. [Google Scholar] [CrossRef]
- Maiga, A.I.; Descamps, D.; Morand-Joubert, L.; Malet, I.; Derache, A.; Cisse, M.; Koita, V.; Akonde, A.; Diarra, B.; Wirden, M.; et al. Resistance-associated mutations to etravirine (TMC-125) in antiretroviral-naive patients infected with non-B HIV-1 subtypes. Antimicrob. Agents Chemother. 2010, 54, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Geretti, A.M.; Zahonero, N.; Booth, C.; Strang, A.; Soriano, V.; De Mendoza, C. Integrase variability and susceptibility to HIV integrase inhibitors: impact of subtypes, antiretroviral experience and duration of HIV infection. J. Antimicrob. Chemother. 2010, 65, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, G.S.; Badiane, N.M.; Hawes, S.E.; Fortes, L.; Toure, M.; Ndour, C.T.; Starling, A.K.; Traore, F.; Sall, F.; Wong, K.G.; et al. Emergence of multiclass drug-resistance in HIV-2 in antiretroviral-treated individuals in Senegal: Implications for HIV-2 treatment in resouce-limited West Africa. Clin. Infect. Dis. 2009, 48, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Ntemgwa, M.L.; d’Aquin Toni, T.; Brenner, B.G.; Camacho, R.J.; Wainberg, M.A. Antiretroviral drug resistance in human immunodeficiency virus type 2. Antimicrob. Agents Chemother. 2009, 53, 3611–3619. [Google Scholar] [CrossRef]
- Nettles, R.E.; Kieffer, T.L.; Kwon, P.; Monie, D.; Han, Y.; Parsons, T.; Cofrancesco, J.Jr.; Gallant, J.E.; Quinn, T.C.; Jackson, B.; Flexner, C.; Carson, K.; Ray, S.; Persaud, D.; Siliciano, R.F. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. JAMA 2005, 293, 817–829. [Google Scholar] [CrossRef]
- Lima, V.; Harrigan, R.; Montaner, J.S. Increased reporting of detectable plasma HIV-1 RNA levels at the critical threshold of 50 copies per milliliter with the Taqman assay in comparison to the Amplicor assay. J. Acquir. Immune Defic. Syndr. 2009, 51, 3–6. [Google Scholar] [CrossRef]
- Smit, E.; Bhattacharya, S.; Osman, H.; Taylor, S. Increased frequency of HIV-1 viral load blip rate observed after switching from Roche Cobas Amplicor to Cobas Taqman assay. J. Acquir. Immune Defic. Syndr. 2009, 51, 364–365. [Google Scholar] [CrossRef]
- Yao, J.D.; Germer, J.J.; Damond, F.; Roquebert, B.; Descamps, D. Plasma load discrepancies between the Roche Cobas Amplicor human immunodeficiency virus type 1 (HIV-1) Monitor version 1.5 and Roche Cobas AmpliPrep/Cobas TaqMan HIV-1 assays. J. Clin. Microbiol. 2008, 46, 834, author reply 834. [Google Scholar] [CrossRef]
- Manavi, K. The significance of low-level plasma HIV viral load on COBAS TaqMan HIV-1 assays for patients with undetectable plasma viral load on COBAS Amplicor monitor version 1.5. HIV Clin. Trials 2008, 9, 283–286. [Google Scholar] [CrossRef]
- Parikh, U.M.; Zelina, S.; Sluis-Cremer, N.; Mellors, J.W. Molecular mechanisms of bidirectional antagonism between K65R and thymidine analog mutations in HIV-1 reverse transcriptase. AIDS 2007, 21, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- AIDSINFO. Available online: http://www.aidsinfo.nih.gov/ (accessed on 21 February 2011).
- Dykes, C.; Najjar, J.; Bosch, R.J.; Wantman, M.; Furtado, M.; Hart, S.; Hammer, S.M.; Demeter, L.M. Detection of drug-resistant minority variants of HIV-1 during virologic failure of indinavir, lamivudine, and zidovudine. J. Infect. Dis. 2004, 189, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Li, J.F.; Wei, X.; Lipscomb, J.; Bennett, D.; Brant, A.; Cong, M.E.; Spira, T.; Shafer, R.W.; Heneine, W. Simple PCR assays improve the sensitivity of HIV-1 subtype B drug resistance testing and allow linking of resistance mutations. PLoS ONE 2007, 2, e638. [Google Scholar] [CrossRef]
- Palmer, S.; Boltz, V.; Martinson, N.; Maldarelli, F.; Gray, G.; McIntyre, J.; Mellors, J.; Morris, L.; Coffin, J. Persistence of nevirapine-resistant HIV-1 in women after single-dose nevirapine therapy for prevention of maternal-to-fetal HIV-1 transmission. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7094–7099. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T.Jr.; Dewar, R.L.; Metcalf, J.A.; Hammer, S.; Mellors, J.W.; Coffin, J.M. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef]
- Johnson, J.A.; Geretti, A.M. Low-frequency HIV-1 drug resistance mutations can be clinically significant but must be interpreted with caution. J. Antimicrob. Chemother. 2010, 65, 1322–1326. [Google Scholar] [CrossRef]
- Geretti, A.M.; Fox, Z.V.; Booth, C.L.; Smith, C.J.; Phillips, A.N.; Johnson, M.; Li, J.F.; Heneine, W.; Johnson, J.A. Low-frequency K103N strengthens the impact of transmitted drug resistance on virologic responses to first-line efavirenz or nevirapine-based highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2009, 52, 569–573. [Google Scholar] [CrossRef]
- Johnson, J.A.; Li, J.F.; Wei, X.; Lipscomb, J.; Irlbeck, D.; Craig, C.; Smith, A.; Bennett, D.E.; Monsour, M.; Sandstrom, P.; Lanier, E.R.; Heneine, W. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naive populations and associate with reduced treatment efficacy. PLoS Med. 2008, 5, e158. [Google Scholar] [CrossRef]
- Paredes, R.; Lalama, C.M.; Ribaudo, H.J.; Schackman, B.R.; Shikuma, C.; Giguel, F.; Meyer, W.A., 3rd; Johnson, V.A.; Fiscus, S.A.; D’Aquila, R.T.; Gulick, R.M.; Kuritzkes, D.R. Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. J. Infect. Dis. 2010, 201, 662–671. [Google Scholar] [CrossRef]
- Arribas, J.R.; Horban, A.; Gerstoft, J.; Fatkenheuer, G.; Nelson, M.; Clumeck, N.; Pulido, F.; Hill, A.; van Delft, Y.; Stark, T.; Moecklinghoff, C. The MONET trial: darunavir/ritonavir with or without nucleoside analogues, for patients with HIV RNA below 50 copies/mL. AIDS 2010, 24, 223–230. [Google Scholar] [CrossRef]
- Boltz, V.; Lockman, Y.Z.S; Hong, F.; Halvas, E.; McIntyre, J.; Currier, J.; Hughes, M.; Coffin, J.; Mellors, J. ; A5208 OCTANE Study Team. In NNRTI-resistant Variants Detected by Allele-specific PCR Predict Outcome of NVP-containing ART in Women with Prior Exposure to sdNVP: Results from the OCTANE/A5208 Study. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, San Francisco, CA, USA, 16–19 February 2010. [Google Scholar]
- Hedskog, C.; Mild, M.; Jernberg, J.; Sherwood, E.; Bratt, G.; Leitner, T.; Lundeberg, J.; Andersson, B.; Albert, J. Dynamics of HIV-1 quasispecies during antiviral treatment dissected using ultra-deep pyrosequencing. PLoS ONE 2010, 5, e11345. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, M.; Chiarella, J.; Yang, R.; Schnittman, S.; Wirtz, V.; Uy, J.; Seekins, D.; Krystal, M.; Mancini, M.; McGrath, D.; Simen, B.; Egholm, M.; Kozal, M. Prevalence and clinical significance of HIV drug resistance mutations by ultra-deep sequencing in antiretroviral-naive subjects in the CASTLE study. PLoS ONE 2010, 5, e10952. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Chiarella, J.; Simen, B.B.; Hanczaruk, B.; Egholm, M.; Landry, M.L.; Dieckhaus, K.; Rosen, M.I.; Kozal, M.J. Low-abundance HIV drug-resistant viral variants in treatment-experienced persons correlate with historical antiretroviral use. PLoS ONE 2009, 4, e6079. [Google Scholar] [CrossRef]
- Mitsuya, Y.; Varghese, V.; Wang, C.; Liu, T.F.; Holmes, S.P.; Jayakumar, P.; Gharizadeh, B.; Ronaghi, M.; Klein, D.; Fessel, W.J.; Shafer, R.W. Minority human immunodeficiency virus type 1 variants in antiretroviral-naive persons with reverse transcriptase codon 215 revertant mutations. J. Virol. 2008, 82, 10747–10755. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.; Braverman, M.S.; Taillon, B.E.; Desany, B.; James, I.; Harrigan, P.R.; Lewis, M.; Robertson, D.L. Detection of low-frequency pretherapy chemokine (CXC motif) receptor 4 (CXCR4)-using HIV-1 with ultra-deep pyrosequencing. AIDS 2009, 23, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Martinez, M.A. HIV-1 reverse transcriptase inhibitor resistance mutations and fitness: A view from the clinic and ex vivo. Virus Res 2008, 134, 104–123. [Google Scholar] [CrossRef]
- Hu, Z.; Giguel, F.; Hatano, H.; Reid, P.; Lu, J.; Kuritzkes, D.R. Fitness comparison of thymidine analog resistance pathways in human immunodeficiency virus type 1. J. Virol. 2006, 80, 7020–7027. [Google Scholar] [CrossRef]
- Hu, Z.; Hatano, H.; Hammond, S.P.; Smith, D.; Wild, M.; Gupta, S.; Whitcomb, J.; Kalayjian, R.C.; Gripshover, B.; Kuritzkes, D.R. Virologic characterization of HIV type 1 with a codon 70 deletion in reverse transcriptase. J. Acquir. Immune Defic. Syndr. 2007, 45, 494–500. [Google Scholar] [CrossRef]
- Hu, Z.; Kuritzkes, D.R. Effect of Raltegravir Resistance Mutations in HIV-1 Integrase on Viral Fitness. J. Acquir. Immune Defic. Syndr. 2010, 55, 148–155. [Google Scholar] [CrossRef]
- Lu, J.; Sista, P.; Giguel, F.; Greenberg, M.; Kuritzkes, D.R. Relative replicative fitness of human immunodeficiency virus type 1 mutants resistant to enfuvirtide (T-20). J. Virol. 2004, 78, 4628–4637. [Google Scholar] [CrossRef]
- Paredes, R.; Sagar, M.; Marconi, V.C.; Hoh, R.; Martin, J.N.; Parkin, N.T.; Petropoulos, C.J.; Deeks, S.G.; Kuritzkes, D.R. In vivo fitness cost of the M184V mutation in multidrug-resistant human immunodeficiency virus type 1 in the absence of lamivudine. J. Virol. 2009, 83, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Barbour, J.D.; Hecht, F.M.; Wrin, T.; Segal, M.R.; Ramstead, C.A.; Liegler, T.J.; Busch, M.P.; Petropoulos, C.J.; Hellmann, N.S.; Kahn, J.O.; Grant, R.M. Higher CD4+ T cell counts associated with low viral pol replication capacity among treatment-naive adults in early HIV-1 infection. J. Infect. Dis. 2004, 190, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Fransen, S.; Karmochkine, M.; Huang, W.; Weiss, L.; Petropoulos, C.J.; Charpentier, C. Longitudinal analysis of raltegravir susceptibility and integrase replication capacity of human immunodeficiency virus type 1 during virologic failure. Antimicrob. Agents Chemother. 2009, 53, 4522–4524. [Google Scholar] [CrossRef] [PubMed]
- Picchio, G.R.; Valdez, H.; Sabbe, R.; Landay, A.L.; Kuritzkes, D.R.; Lederman, M.M.; Mosier, D.E. Altered viral fitness of HIV-1 following failure of protease inhibitor-based therapy. J. Acquir. Immune Defic. Syndr. 2000, 25, 289–295. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, H.; Acosta, E.P. Hierarchical Bayesian inference for HIV dynamic differential equation models incorporating multiple treatment factors. Biom J. 2010, 52, 470–486. [Google Scholar] [CrossRef]
- Prosperi, M.C.; Altmann, A.; Rosen-Zvi, M.; Aharoni, E.; Borgulya, G.; Bazso, F.; Sonnerborg, A.; Schulter, E.; Struck, D.; Ulivi, G.; Vandamme, A.M.; Vercauteren, J.; Zazzi, M. Investigation of expert rule bases, logistic regression, and non-linear machine learning techniques for predicting response to antiretroviral treatment. Antivir. Ther. 2009, 14, 433–442. [Google Scholar] [CrossRef]
- Revell, A.D.; Wang, D.; Harrigan, R.; Hamers, R.L.; Wensing, A.M.; Dewolf, F.; Nelson, M.; Geretti, A.M.; Larder, B.A. Modelling response to HIV therapy without a genotype: an argument for viral load monitoring in resource-limited settings. J. Antimicrob. Chemother. 2010, 65, 605–607. [Google Scholar] [CrossRef]
- Swindells, S.; DiRienzo, A.G.; Wilkin, T.; Fletcher, C.V.; Margolis, D.M.; Thal, G.D.; Godfrey, C.; Bastow, B.; Ray, M.G.; Wang, H.; Coombs, R.W.; McKinnon, J.; Mellors, J.W. Regimen simplification to atazanavir-ritonavir alone as maintenance antiretroviral therapy after sustained virologic suppression. JAMA 2006, 296, 806–814. [Google Scholar] [CrossRef]
- Wilkin, T.J.; McKinnon, J.E.; DiRienzo, A.G.; Mollan, K.; Fletcher, C.V.; Margolis, D.M.; Bastow, B.; Thal, G.; Woodward, W.; Godfrey, C.; et al. Regimen simplification to atazanavir-ritonavir alone as maintenance antiretroviral therapy: final 48-week clinical and virologic outcomes. J. Infect. Dis. 2009, 199, 866–871. [Google Scholar] [CrossRef]
Figure 2.
Mutations conferring resistance to nucleotide reverse transcriptase inhibitors (NRTIs) are depicted; multidrug resistance profile (Q151 complex) is indicated and thymidine associated mutations (TAMS) are noted.
Figure 2.
Mutations conferring resistance to nucleotide reverse transcriptase inhibitors (NRTIs) are depicted; multidrug resistance profile (Q151 complex) is indicated and thymidine associated mutations (TAMS) are noted.

Figure 3.
(A) Crystal structure of HIV reverse transcriptase p66/p561 dimer is depicted with locations of four common NNRTI resistance mutations in a hydrophobic pocket within the palm domain noted in yellow. Mutations change the binding characteristics of NNRTI, explaining cross resistance of nevirapine, delavirdine, and efavirenz. Structural data are from [99] and are displayed using RASMOL [100,101]. (B) Mutations conferring resistance to individual NNRTIs. Despite some cross resistance, etravirine has antiviral activity even in the presence of a number of mutations conferring resistance to nevirapine and efavirenz.
Figure 3.
(A) Crystal structure of HIV reverse transcriptase p66/p561 dimer is depicted with locations of four common NNRTI resistance mutations in a hydrophobic pocket within the palm domain noted in yellow. Mutations change the binding characteristics of NNRTI, explaining cross resistance of nevirapine, delavirdine, and efavirenz. Structural data are from [99] and are displayed using RASMOL [100,101]. (B) Mutations conferring resistance to individual NNRTIs. Despite some cross resistance, etravirine has antiviral activity even in the presence of a number of mutations conferring resistance to nevirapine and efavirenz.

Figure 4.
(A) Crystal structure of HIV protease depicting active site residues (yellow) and a series of residues conferring resistance near the active site or at flap domains (circled). Structural data are from [114] and are displayed using RASMOL [100,101]. (B) Chart depicting resistance to HIV-1 protease.
Figure 4.
(A) Crystal structure of HIV protease depicting active site residues (yellow) and a series of residues conferring resistance near the active site or at flap domains (circled). Structural data are from [114] and are displayed using RASMOL [100,101]. (B) Chart depicting resistance to HIV-1 protease.


Figure 5.
Crystal structure of human foamy virus integrase (similar to HIV integrase) complexed with DNA substrate, noting the positions of resistance mutations 143, 148, and 155 relative to the binding site for raltegravir and elvitegravir, and proximity to DNA. Structure is from Cherapenov and coworkers [128] and rendered in RASMOL [100,101].
Figure 5.
Crystal structure of human foamy virus integrase (similar to HIV integrase) complexed with DNA substrate, noting the positions of resistance mutations 143, 148, and 155 relative to the binding site for raltegravir and elvitegravir, and proximity to DNA. Structure is from Cherapenov and coworkers [128] and rendered in RASMOL [100,101].

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Cortez, K.J.; Maldarelli, F. Clinical Management of HIV Drug Resistance. Viruses 2011, 3, 347-378. https://0-doi-org.brum.beds.ac.uk/10.3390/v3040347
AMA Style
Cortez KJ, Maldarelli F. Clinical Management of HIV Drug Resistance. Viruses. 2011; 3(4):347-378. https://0-doi-org.brum.beds.ac.uk/10.3390/v3040347
Chicago/Turabian StyleCortez, Karoll J., and Frank Maldarelli. 2011. "Clinical Management of HIV Drug Resistance" Viruses 3, no. 4: 347-378. https://0-doi-org.brum.beds.ac.uk/10.3390/v3040347