Detection and Molecular Diversity of Spike Gene of Porcine Epidemic Diarrhea Virus in China

Abstract
:1. Introduction
2. Results
2.1. PEDV Detection
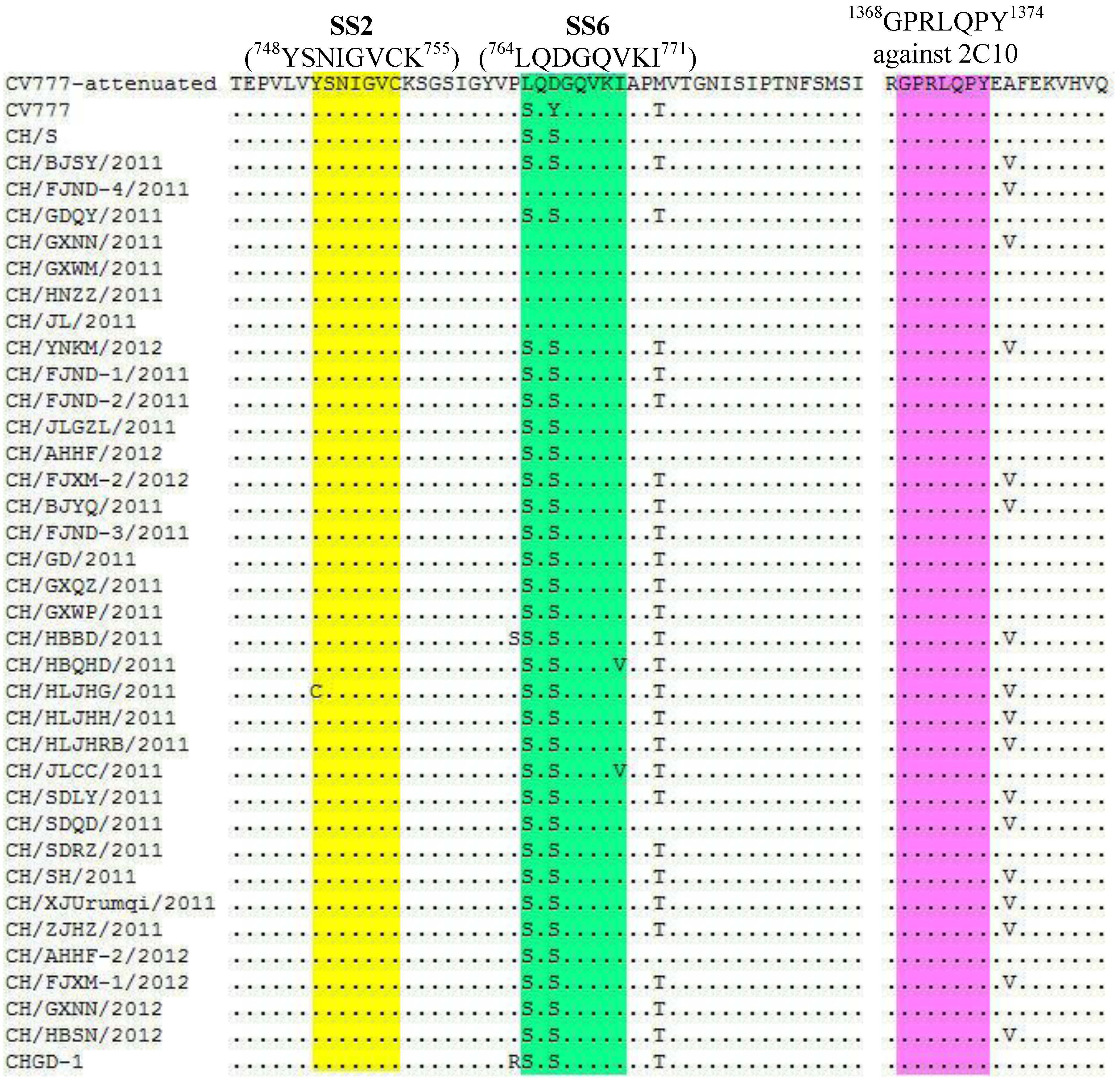
2.2. Nucleotide and Amino Acid Sequence Analysis

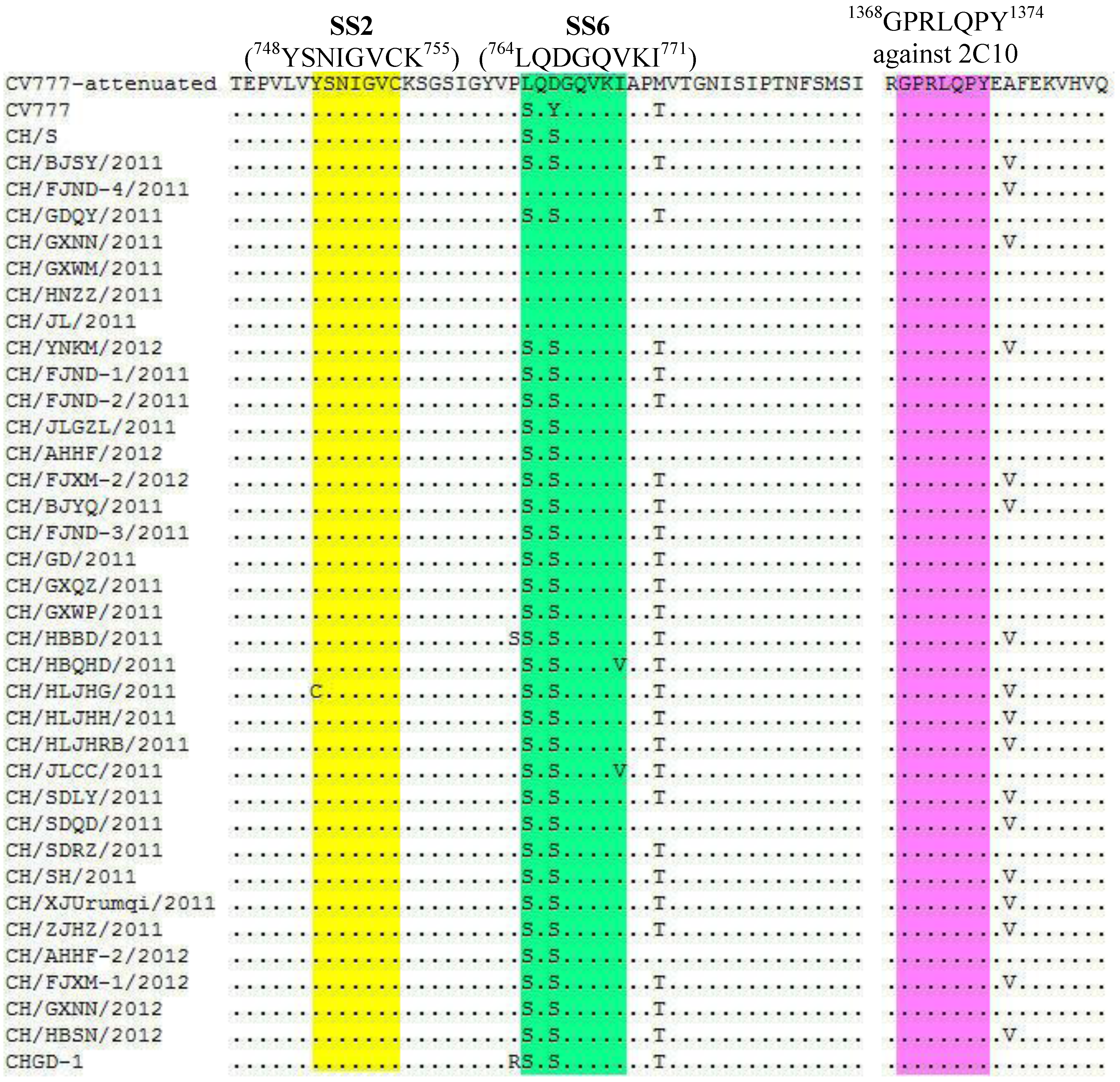{kind=link}
{kind=link}
| Field strains | Abbreviations | Regions | S genes (nt) | S proteins (aa) | Accession numbers |
|---|---|---|---|---|---|
| CH/FJND-2/2011 | FJND-2 | Ningde, Fujian | 4146 | 1381 | JN315706 |
| CH/GDQY/2011 | GDQY | Qingyuan ,Guangdong | 4146 | 1381 | JN601051(S1);JQ638914(S2) |
| CH/GXWM/2011 | GXWM | Wuming, Guangxi | 4146 | 1381 | JN601045(S1);JQ638911(S2) |
| CH/BJSY/2011 | BJSY | Shunyi, Beijing | 4149 | 1382 | JQ638921 |
| CH/FJND-1/2011 | FJND-1 | Ningde, Fujian | 4149 | 1382 | JN543367 |
| CH/FJND-4/2011 | FJND-4 | Ningde, Fujian | 4149 | 1382 | JN601044(S1);JQ638506(S2) |
| CH/GXNN/2011 | GXNN-1 | Nanning, Guangxi | 4149 | 1382 | JN601049(S1);JQ638912(S2) |
| CH/HNZZ/2011 | HNZZ | Zhengzhou, Henan | 4149 | 1382 | JN601050(S1);JQ638512(S2) |
| CH/JL/2011 | JL | Jilin | 4149 | 1382 | JQ638924 |
| CH/AHHF/2012 | AHHF | Hefei, Anhui | 4149 | 1382 | JX018181 |
| CH/YNKM/2012 | YNKM | Kunming, Yunnan | 4149 | 1382 | JX018180 |
| CH/JLGZL/2011 | JLGZL | Siping, Jilin | 4152 | 1383 | JQ638923 |
| CH/FJXM-2/2012 | FJXM-2 | Xiamen, Fujian | 4152 | 1383 | JX070672 |
| CH/GD/2011 | GD | Guangdong | 4158 | 1385 | JQ638915 |
| CH/GXNN/2012 | GXNN-2 | Nanning, Guangxi | 4158 | 1385 | JX018179 |
| CH/BJYQ/2011 | BJYQ | Yanqing, Bejing | 4161 | 1386 | JN601048(S1); JQ305101(S2) |
| CH/GXQZ/2011 | GXQZ | Qinzhou, Guangxi | 4161 | 1386 | JN641881(S1);JQ638913(S2) |
| CH/GXWP/2011 | GXWP | Nanning, Guangxi | 4161 | 1386 | JN641879(S1);JQ638513(S2) |
| CH/HBQHD/2011 | HBQHD | Qihuangdao, Hebei | 4161 | 1386 | JQ638922 |
| CH/HLJHG/2011 | HLJHG | Hegang, Heilongjiang | 4161 | 1386 | JN601046(S1);JQ638508(S2) |
| CH/HLJHH/2011 | HLJHH | Heihe, Heilongjiang | 4161 | 1386 | JQ638916 |
| CH/HLJHRB/2011 | HLJHRB | Harbin, Heilongjiang | 4161 | 1386 | JN711456(S1);JQ638507(S2) |
| CH/JLCC/2011 | JLCC | Chengchun, Jilin | 4161 | 1386 | JQ638920 |
| CH/SDLY/2011 | SDLY | Linyi, Shandong | 4161 | 1386 | JQ638917 |
| CH/SDQD/2011 | SDQD | Qingdao, Shandong | 4161 | 1386 | JQ638919 |
| CH/SDRZ/2011 | SDRZ | Rizhao, Shandong | 4161 | 1386 | JN671916(S1);JQ638505(S2) |
| CH/SH/2011 | SH | Shanghai | 4161 | 1386 | JN711457(S1);JQ638511(S2) |
| CH/ZJHZ/2011 | ZJHZ | Hangzhou, Zhejiang | 4161 | 1386 | JN641880(S1);JQ638509(S2) |
| CH/XJUrumqi/2011 | XJUrumqi | Urumqi, Xinjiang | 4161 | 1386 | JN601047(S1);JQ638510(S2) |
| CH/AHHF-2/2012 | AHHF-2 | Hefei, Anhui | 4161 | 1386 | JX018182 |
| CH/FJXM-1/2012 | FJXM-1 | Xiamen, Fujian | 4161 | 1386 | JX070671 |
| CH/HBSN/2012 | HBSN | Suning, Hebei | 4161 | 1386 | JX018183 |
| CH/HBBD/2011 | HBBD | Baoding, Hebei | 4170 | 1389 | JQ638918 |
| Field strains | Amino acids in COE of CV777 attenuated strain | Total | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 500S | 505I | 509T | 520N | 526A | 530H | 537A | 543N | 546S | 558T | 572K | 575D | 576S | 603G | 610F | 614E | 615F | 617S | 618G | 629T | 632E | ||
| BJSY, FJND-4 | 0 | |||||||||||||||||||||
| GDQY, GXNN-1 | 0 | |||||||||||||||||||||
| HNZZ, JL | 0 | |||||||||||||||||||||
| NKM | 0 | |||||||||||||||||||||
| GXWM | A | 1 | ||||||||||||||||||||
| JLGZL, HBSN | S | S | S | 3 | ||||||||||||||||||
| FJXM-1,GXNN-2 | T | S | S | 3 | ||||||||||||||||||
| FJXM-2 | S | V | S | S | 4 | |||||||||||||||||
| GXQZ, JLCC | T | S | N | S | 4 | |||||||||||||||||
| SDLY | A | S | S | S | 4 | |||||||||||||||||
| FJND-1, -2 | S | P | S | S | G | 5 | ||||||||||||||||
| BJYQ | S | P | S | S | G | 5 | ||||||||||||||||
| SDRZ | T | I | S | N | S | 5 | ||||||||||||||||
| XJUrumqi | T | S | R | S | S | 5 | ||||||||||||||||
| ZJHZ | T | S | N | S | I | 5 | ||||||||||||||||
| AHHF, AHHF-2 | S | S | N | N | T | S | 6 | |||||||||||||||
| GXWP | S | S | T | S | D | Y | 6 | |||||||||||||||
| HBBD | T | G | S | S | L | A | 6 | |||||||||||||||
| HBQHD | T | R | S | N | S | G | 6 | |||||||||||||||
| SDQD | T | S | L | S | S | D | 6 | |||||||||||||||
| GD | I | S | S | T | S | D | Y | 7 | ||||||||||||||
| HLJHG | T | R | S | S | N | S | G | 7 | ||||||||||||||
| HLJHH, HLJHRB | I | S | S | T | S | D | Y | 7 | ||||||||||||||
| SH | I | S | S | T | S | D | Y | G | 8 | |||||||||||||
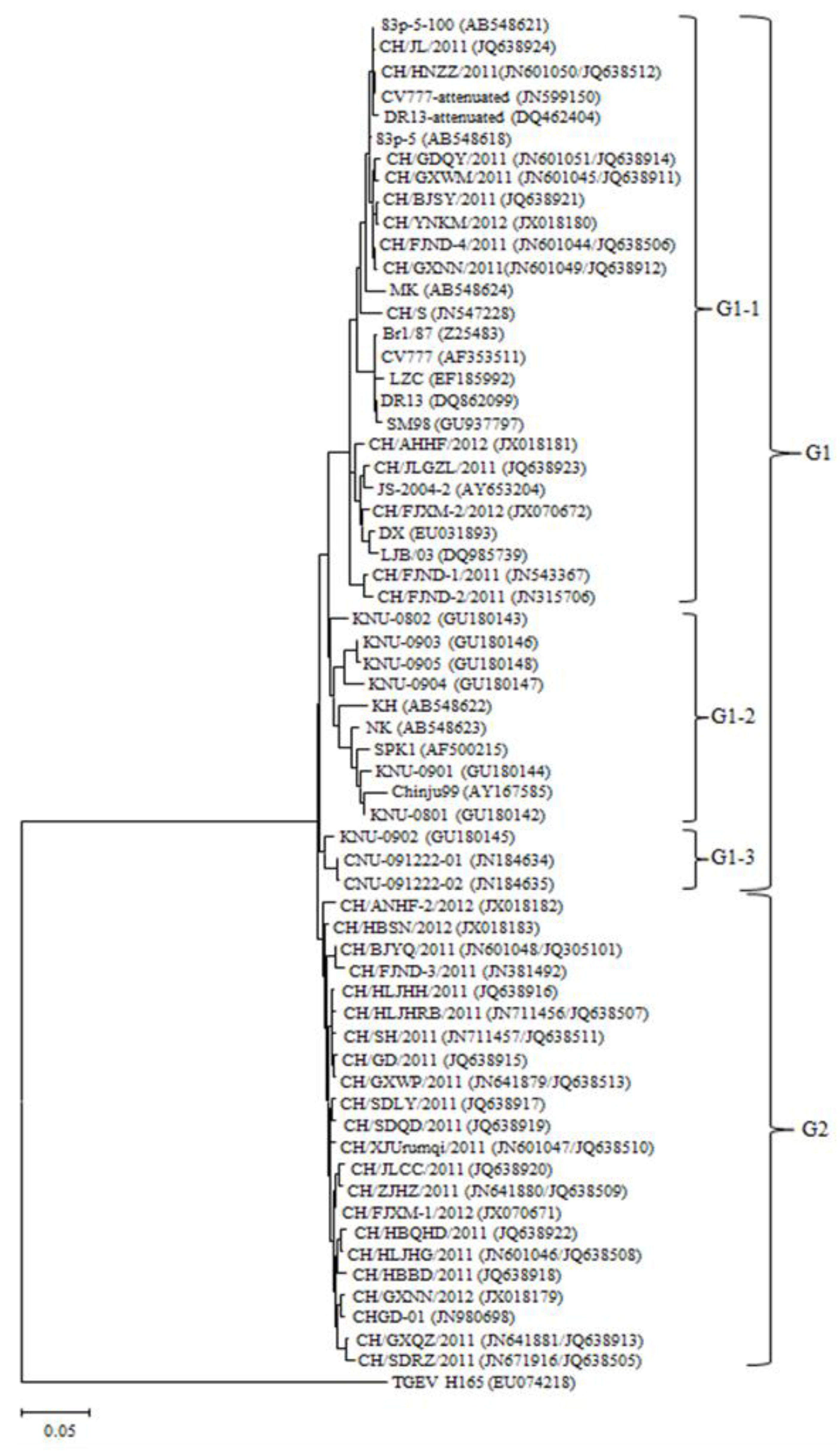
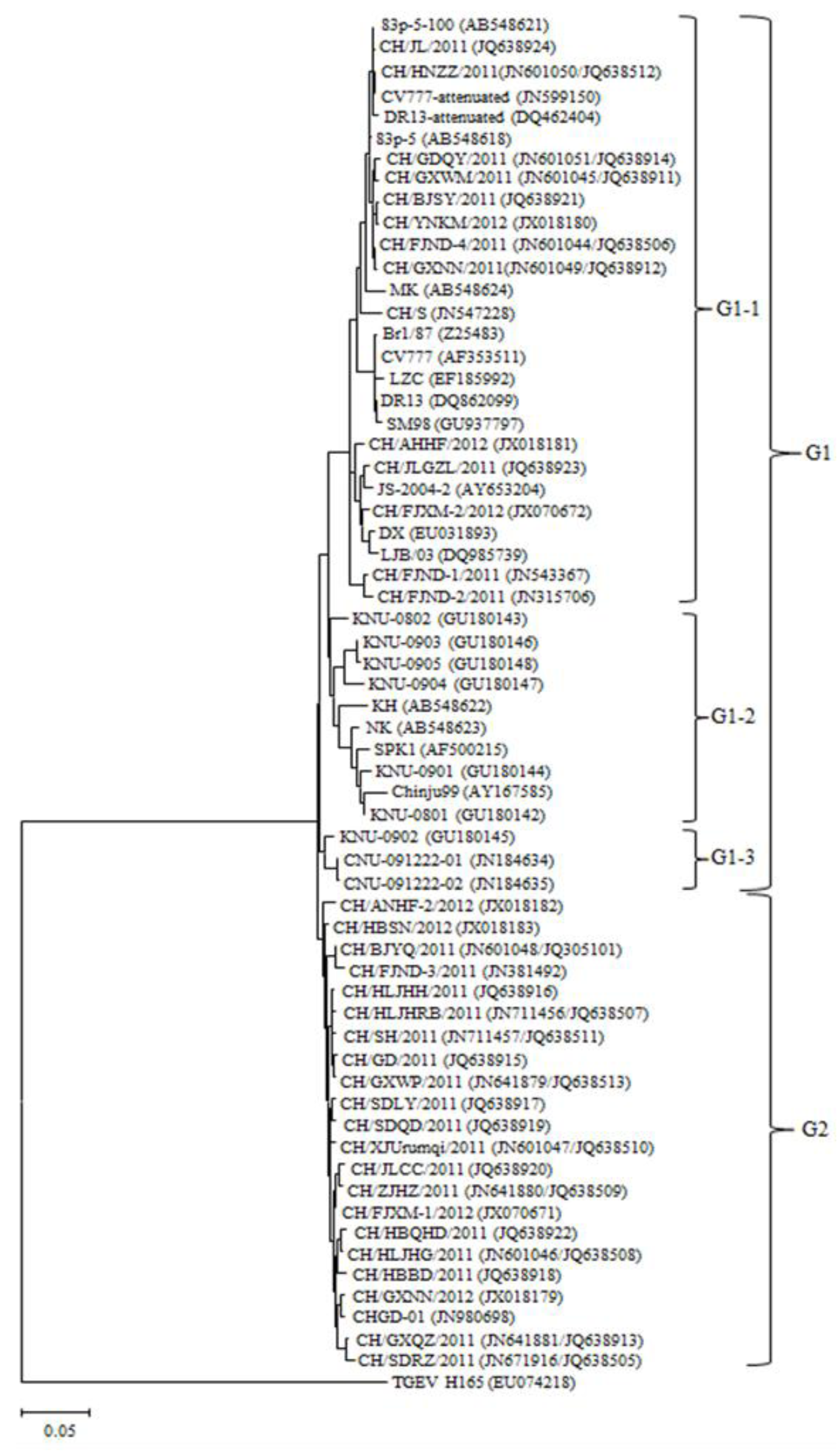
2.3. Phylogenetic Analysis
2.4. Sequence Homology Analysis
| Groups and reference strains | Percentage identity (%) a | |||||
|---|---|---|---|---|---|---|
| G1-1 | G2 | CV777-attenuated | CH/S | CHGD-1 | ||
| Percentage identity (%) b | G1-1 | 95.6–99.9 c 94.9–99.8 | 93.5–97.1 | 96.0–99.9 | 95.4–96.7 | 93.6–95.7 |
| G2 | 92.3–96.5 | 96.2–99.7 d 96.7–99.7 | 93.6–95.4 | 93.5–94.2 | 97.1–99.3 | |
| CV777-attenuated | 95.9–100 | 92.3–94.4 | *** | 96.7 | 93.6 | |
| CH/S | 94.6–96.2 | 92.9–93.7 | 96.2 | *** | 93.3 | |
| CHGD-1 | 92.7–95.1 | 96.6–98.9 | 92.7 | 93.0 | *** | |
3. Discussion
4. Experimental
4.1. Clinical Samples Detection
4.2. Sequencing of S Gene
4.3. Multiple Alignments and Phylogenetic Analysis
| Reference strains | Countries | S genes (nt) | S proteins (aa) | Accession no. |
|---|---|---|---|---|
| CV777 | Belgium | 4152 | 1383 | AF353511 |
| Br1/87 | British | 4152 | 1383 | Z25483 |
| CH/S | China | 4152 | 1383 | JN547228 |
| LZC | China | 4152 | 1383 | EF185992 |
| JS-2004-02 | China | 4152 | 1383 | AY653204 |
| DX | China | 4152 | 1383 | EU031893 |
| LJB/03 | China | 4152 | 1383 | DQ985739 |
| CV777 attenuated | China | 4149 | 1382 | JN599150 |
| CHGD-1 | China | 4158 | 1385 | JN980698 |
| CH/FJND-3/2011 | China | 4161 | 1386 | JQ282909 |
| Chinju99 | South Korea | 4152 | 1383 | AY167585 |
| DR13 | South Korea | 4152 | 1383 | DQ862099 |
| DR13 attenuated | South Korea | 4149 | 1382 | DQ462404 |
| SM98 | South Korea | 4143 | 1380 | GU937797 |
| SPK1 | South Korea | 4161 | 1386 | AF500215 |
| KNU-0801 | South Korea | 4161 | 1386 | GU180142 |
| KNU-0802 | South Korea | 4161 | 1386 | GU180143 |
| KNU-0901 | South Korea | 4161 | 1386 | GU180144 |
| KNU-0902 | South Korea | 4161 | 1386 | GU180145 |
| KNU-0903 | South Korea | 4161 | 1386 | GU180146 |
| KNU-0904 | South Korea | 4161 | 1386 | GU180147 |
| KNU-0905 | South Korea | 4161 | 1386 | GU180148 |
| CNU-091222-01 | South Korea | 4161 | 1386 | JN184634 |
| CNU-091222-02 | South Korea | 4161 | 1386 | JN184635 |
| 83p-5 | Japan | 4152 | 1383 | AB548618 |
| 83p-5-100 | Japan | 4149 | 1382 | AB548621 |
| KH | Japan | 4164 | 1387 | AB548622 |
| NK | Japan | 4176 | 1391 | AB548623 |
| MK | Japan | 4152 | 1383 | AB548624 |
| TGEV H165 | China | 4347 | 1448 | EU074218 |
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pensaert, M.B.; DeBouck, P. A new coronavirus-like particles associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef]
- Oldham, J. Letter to the editor. Pig Farming 1972, 10, 72–73. [Google Scholar]
- Pensaert, M.B.; Callebaut, P.; DeBouck, P. Porcine epidemic diarrhea (PED) caused by a coronavirus: Present knowledge. Proc. Congr. Int. Pig. Vet. Soc. 1982, 7, 52. [Google Scholar]
- Xuan, H.; Xing, D.; Wang, D.; Zhu, W.; Zhao, F.; Gong, H. Study on the culture of porcine epidemic diarrhea virus adapted to fetal porcine intestine primary cell monolayer. Chin. J. Vet. Sci. 1984, 4, 202–208. [Google Scholar]
- Sun, R.Q.; Cai, R.J.; Chen, Y.Q.; Liang, P.S.; Chen, D.K.; Song, C.X. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 2011, 18, 161–163. [Google Scholar]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350–1353. [Google Scholar]
- Sánchez, C.M.; Gebauer, F.; Suñé, C.; Mendez, A.; Dopazo, J.; Enjuanes, L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 1992, 190, 92–105. [Google Scholar] [CrossRef]
- Paton, D.; Lowings, P. Discrimination between transmissible gastroenteritis virus isolates. Arch. Virol. 1997, 142, 1703–1711. [Google Scholar] [CrossRef]
- Kim, S.J.; Han, J.H.; Kwon, H.M. Partial sequence of the spike glycoprotein gene of transmissible gastroenteritis viruses isolated in Korea. Vet. Microbiol. 2003, 94, 195–206. [Google Scholar] [CrossRef]
- Park, S.J.; Moon, H.J.; Yang, J.S.; Lee, C.S.; Song, D.S.; Kang, B.K.; Park, B.K. Cloning and further sequence analysis of the spike gene of attenuated porcine epidemic diarrhea virus DR13. Virus Genes 2007, 35, 55–64. [Google Scholar] [CrossRef]
- Puranaveja, S.; Poolperm, P.; Lertwatcharasarakul, P.; Kesdaengsakonwut, S.; Boonsoongnern, A.; Urairong, K.; Kitikoon, P.; Choojai, P.; Kedkovid, R.; Teankum, K.; et al. Chinese-like strain of porcine epidemic diarrhea virus, Thailand. Emerg. Infect. Dis. 2009, 15, 1112–1115. [Google Scholar] [CrossRef]
- Lee, D.K.; Park, C.K.; Kim, S.H.; Lee, C. Heterogeneity in spike protein genes of porcine epidemic diarrhea viruses isolated in Korea. Virus Res. 2010, 149, 175–182. [Google Scholar] [CrossRef]
- Cruz, D.J.; Kim, C.J.; Shin, H.J. Phage-displayed peptides having antigenic similarities with porcine epidemic diarrhea virus (PEDV) neutralizing epitopes. Virology 2006, 354, 28–34. [Google Scholar] [CrossRef]
- Chang, S.H.; Bae, J.L.; Kang, T.J.; Kim, J.; Chung, G.H.; Lim, C.W.; Laude, H.; Yang, M.S.; Jang, Y.S. Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol. Cells 2002, 14, 295–299. [Google Scholar]
- Sun, D.; Feng, L.; Shi, H.; Chen, J.; Cui, X.; Chen, H.; Liu, S.; Tong, Y.; Wang, Y.; Tong, G. Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Vet. Microbiol. 2008, 131, 73–81. [Google Scholar] [CrossRef]
- Wang, C.; Chen, J.; Shi, H.; Qiu, H.J.; Xue, F.; Liu, S.; Liu, C.; Zhu, Y.; Almazán, F.; Enjuanes, L.; et al. Rapid differentiation of vaccine strain and Chinese field strains of transmissible gastroenteritis virus by restriction fragment length polymorphism of the N gene. Virus Genes 2010, 41, 47–58. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Shi, D.; Shi, H.; Zhang, X. ; Feng, L. Complete genome sequence of a porcine epidemic diarrhea virus variant. J. Virol. 2012, 86. [Google Scholar] [CrossRef]
- Park, S.J.; Song, D.S.; Ha, G.W.; Park, B.K. Sequence analysis of the partial spike glycoprotein gene of porcine epidemic diarrhea viruses isolated in Korea. Virus Genes 2007, 35, 321–332. [Google Scholar] [CrossRef]
- Sato, T.; Takeyama, N.; Katsumata, A.; Tuchiya, K.; Kodama, T.; Kusanagi, K. Mutations in the spike gene of porcine epidemic diarrhea virus associated with growth adaptation in vitro and attenuation of virulence in vivo. Virus Genes 2011, 43, 72–78. [Google Scholar] [CrossRef]
- Kocherhans, R.; Bridgen, A.; Ackermann, M.; Tobler, K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes 2001, 23, 137–144. [Google Scholar] [CrossRef]
- Duarte, M.; Laude, H. Sequence of the spike protein of porcine epidemic diarrhea virus. J. Gen. Virol. 1994, 75, 1195–1200. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.; Shi, H.; Qiu, H.J.; Liu, S.; Shi, D.; Zhang, X.; Feng, L. Complete genome sequence of a Chinese virulent porcine epidemic diarrhea virus strain. J. Virol. 2011, 85, 11538–11539. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.; Shi, H.; Qiu, H.J.; Liu, S.; Chen, X.; Zhang, Z.; Feng, L. Molecular epidemiology of porcine epidemic diarrhea virus in China. Arch. Virol. 2010, 155, 1471–1476. [Google Scholar] [CrossRef]
- DNAStar package, version 7.1.0. Lasergene; DNAStar, Inc.: Madison, WI, USA, 2006.
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Med. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, J.; Liu, X.; Shi, D.; Shi, H.; Zhang, X.; Li, C.; Chi, Y.; Feng, L. Detection and Molecular Diversity of Spike Gene of Porcine Epidemic Diarrhea Virus in China. Viruses 2013, 5, 2601-2613. https://0-doi-org.brum.beds.ac.uk/10.3390/v5102601
Chen J, Liu X, Shi D, Shi H, Zhang X, Li C, Chi Y, Feng L. Detection and Molecular Diversity of Spike Gene of Porcine Epidemic Diarrhea Virus in China. Viruses. 2013; 5(10):2601-2613. https://0-doi-org.brum.beds.ac.uk/10.3390/v5102601
Chicago/Turabian StyleChen, Jianfei, Xiaozhen Liu, Da Shi, Hongyan Shi, Xin Zhang, Changlong Li, Yanbin Chi, and Li Feng. 2013. "Detection and Molecular Diversity of Spike Gene of Porcine Epidemic Diarrhea Virus in China" Viruses 5, no. 10: 2601-2613. https://0-doi-org.brum.beds.ac.uk/10.3390/v5102601