
Genome Characterization, Prevalence and Distribution of a Macula-Like Virus from Apis mellifera and Varroa destructor

 ,
, 
 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Origins, RNA Purification and cDNA Synthesis
2.2. PCR and qPCR Assays
2.3. Cloning, Sequencing, and Assembly
2.4. Phylogenetic and Variability Analyses
2.5. Statistical Analyses
3. Results
3.1. Discovery
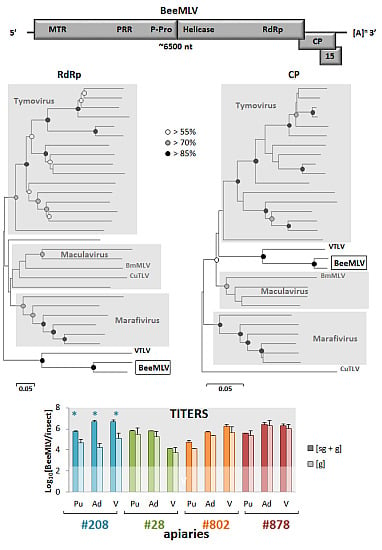
3.2. Genome Sequencing and Analysis


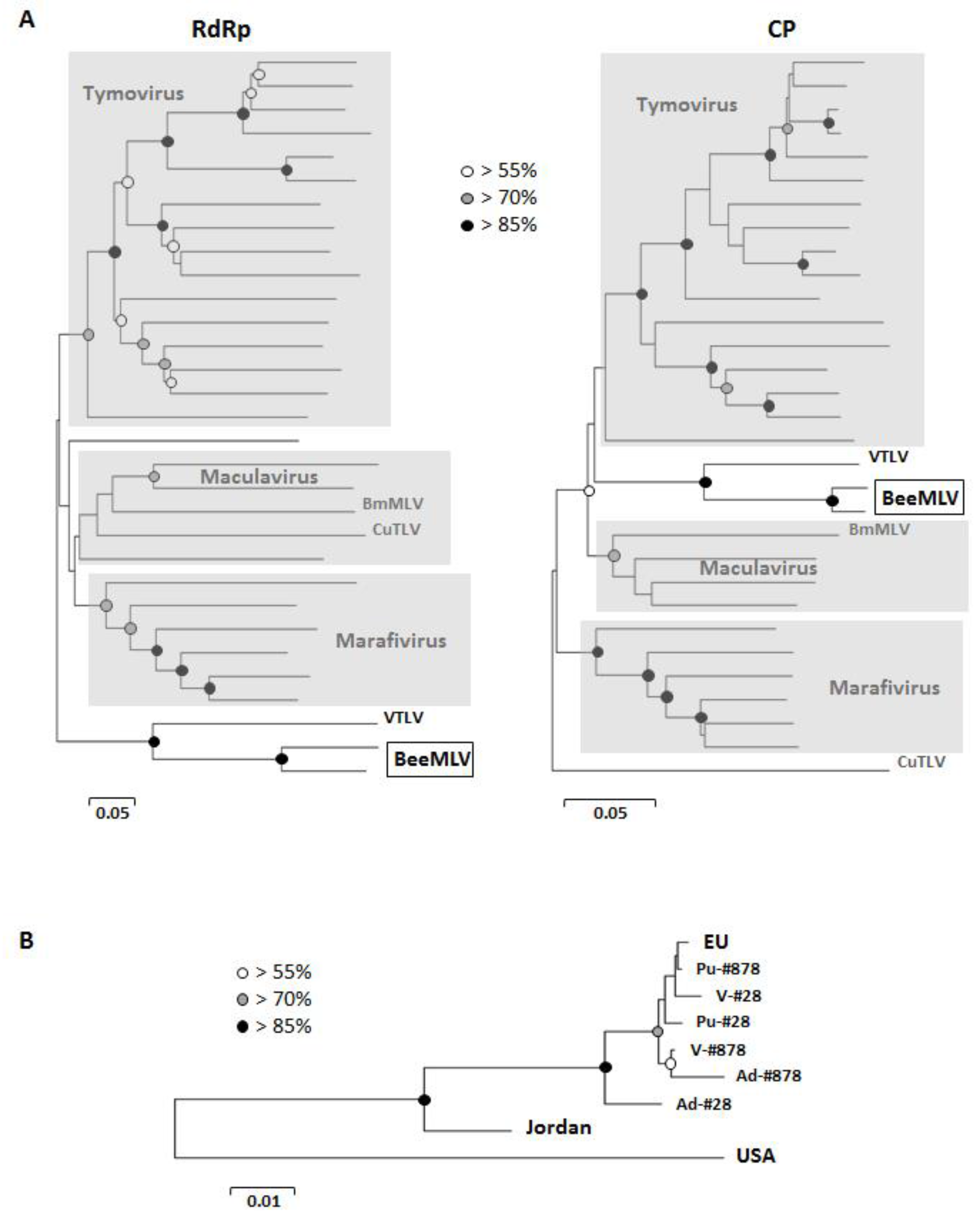
3.3. Phylogenetic Analyses
3.4. Reference Collection Analysis
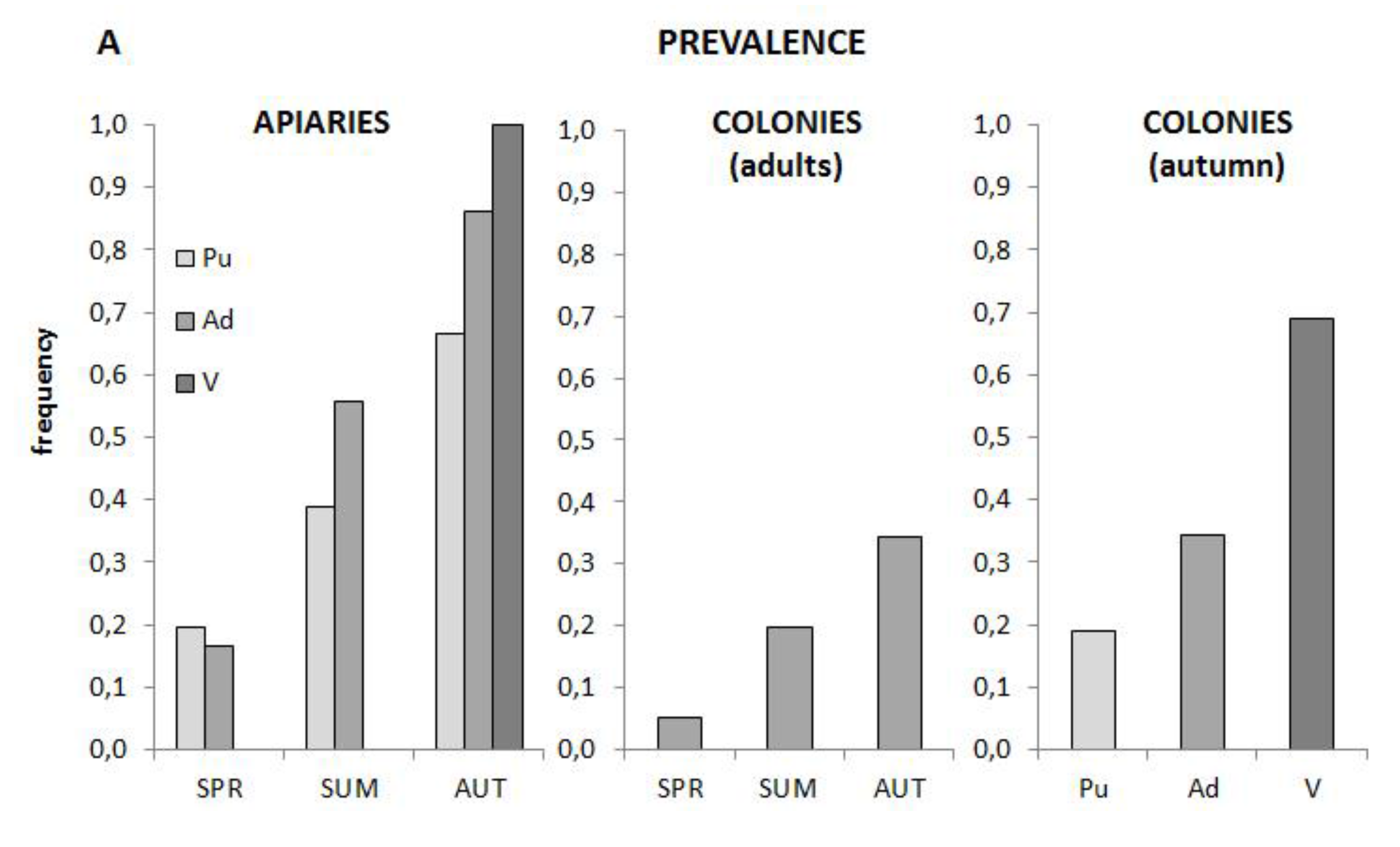
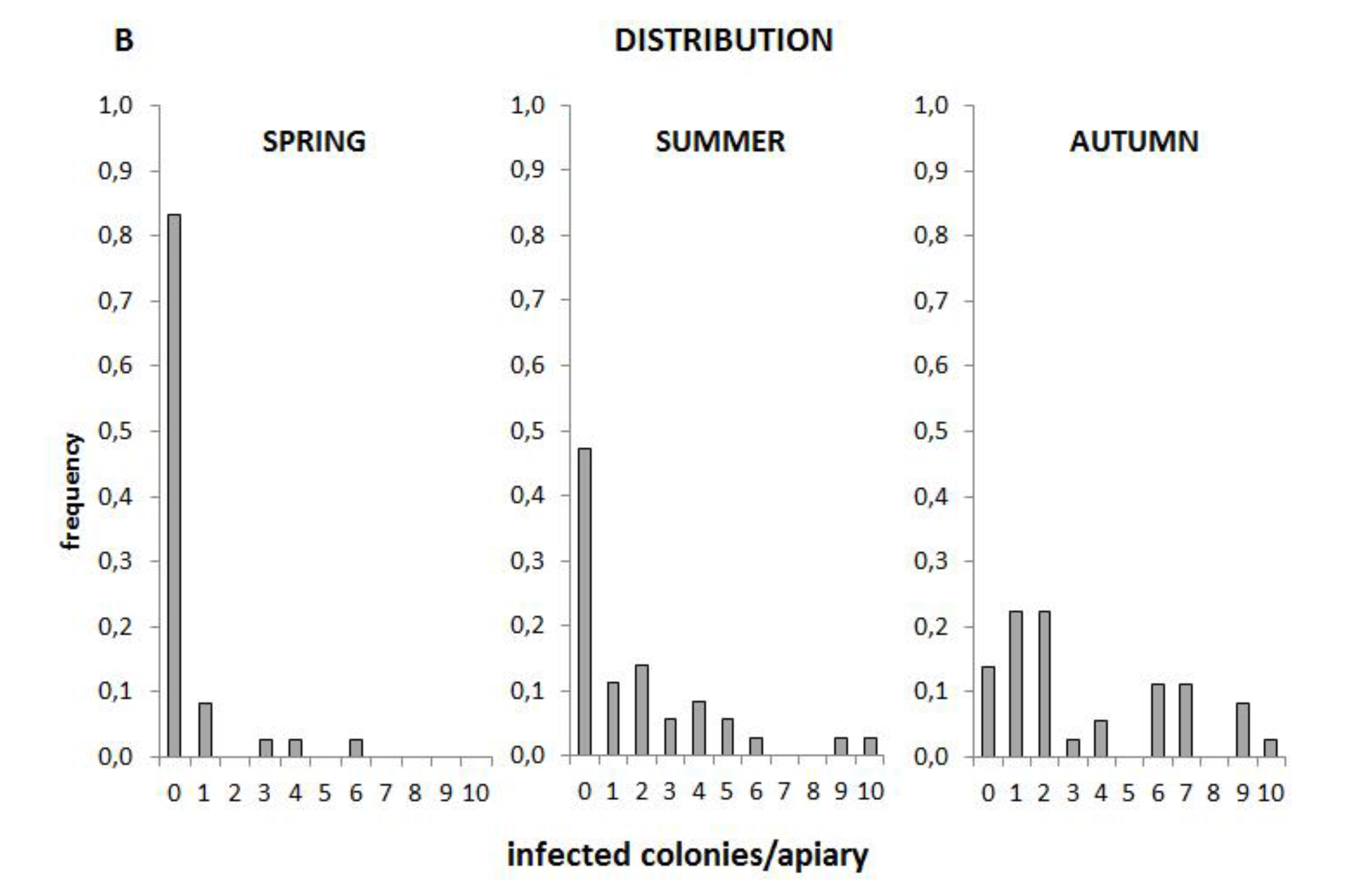
3.5. Prevalence and Distribution


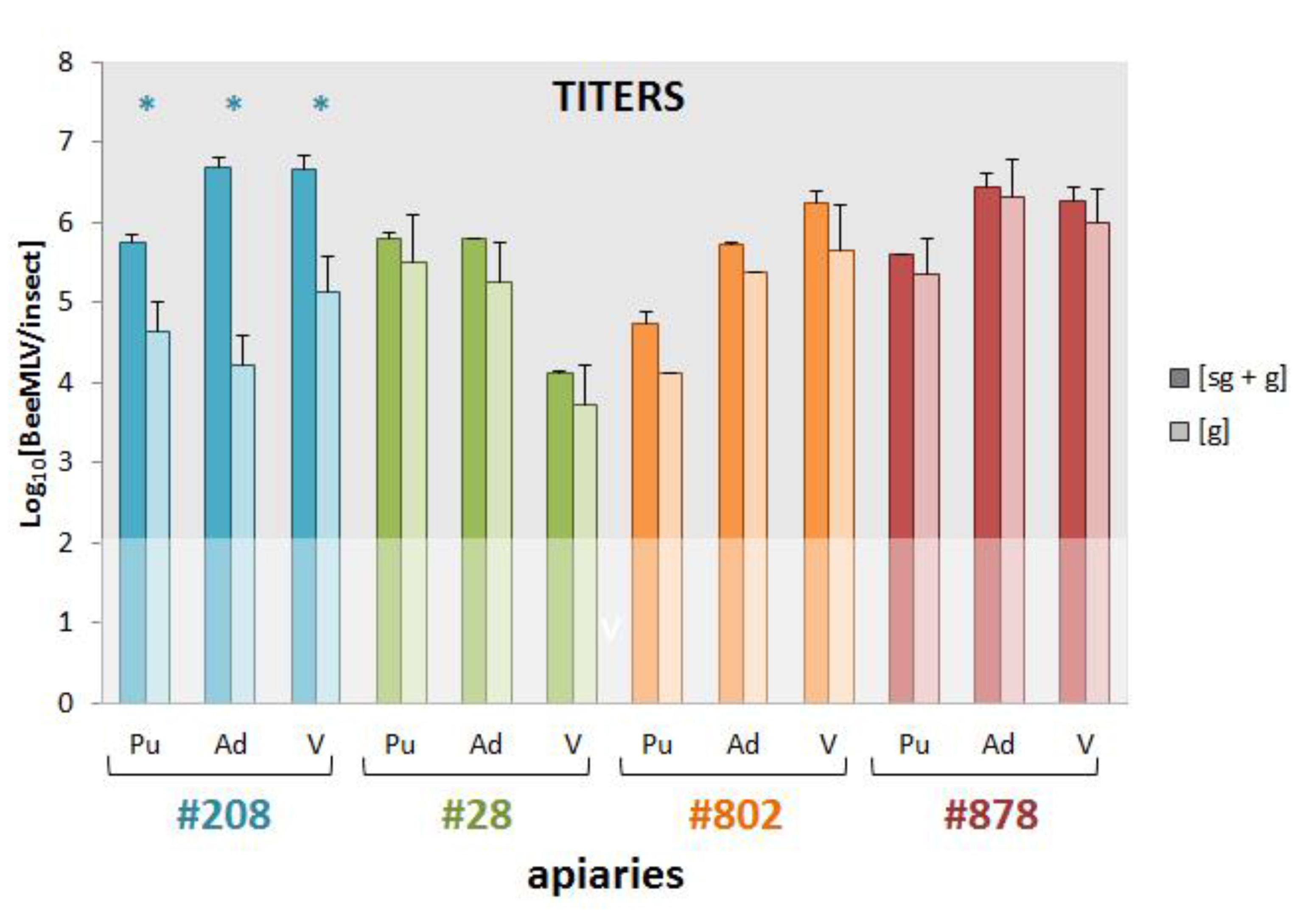
3.6. Genomic and Sub-Genomic BeeMLV Titers

4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bailey, L.; Ball, B.V. Honey Bee Pathology, 2nd ed.; Academic Press: London, UK, 1991; pp. 1–193. [Google Scholar]
- de Miranda, J.R.; Bailey, L.; Ball, B.V.; Blanchard, P.; Budge, G.; Chejanovsky, N.; Chen, Y.P.; Gauthier, L.; Genersch, E.; de Graaf, D.; et al. Standard methods for virus research in Apis mellifera. In The COLOSS BEEBOOK, Volume II: Standard Methods for Apis mellifera Pest and Pathogen Research; Dietemann, V., Ellis, J.D., Neumann, P., Eds.; IBRA: Treforest, UK, 2013; Chapter 13; pp. 1–55. [Google Scholar]
- Allen, M.F.; Ball, B.V. Characterisation and serological relationships of strains of Kashmir bee virus. Ann. Appl. Biol. 1995, 126, 471–484. [Google Scholar] [CrossRef]
- de Miranda, J.R.; Cordoni, G.; Budge, G. The acute bee paralysis virus–Kashmir bee virus–Israeli acute paralysis virus complex. J. Invertebr. Pathol. 2010, 103, S30–S47. [Google Scholar] [CrossRef] [PubMed]
- de Miranda, J.R.; Genersch, E. Deformed wing virus. J. Invertebr. Pathol. 2010, 103, S48–S61. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, G.; de Miranda, J.R.; Boniotti, M.B.; Cameron, C.E.; Lavazza, A.; Capucci, L.; Camazine, S.M.; Rossi, C. Molecular and biological characterization of deformed wing virus of honeybees (Apis mellifera L.). J. Virol. 2006, 80, 4998–5009. [Google Scholar] [CrossRef] [PubMed]
- Tentcheva, D.; Gauthier, L.; Zappulla, N.; Dainat, B.; Cousserans, F.; Colin, M.E; Bergoin, M. Prevalence and seasonal variations of six bee viruses in Apis mellifera L. and Varroa destructor mite populations in France. Appl. Environ. Microbiol. 2004, 70, 7185–7191. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Tentcheva, D.; Tournaire, M.; Dainat, B.; Cousserans, F.; Colin, M.E; Bergoin, M. Viral load estimation in asymptomatic honey bee colonies using the quantitative RT-PCR technique. Apidologie 2007, 38, 426–435. [Google Scholar] [CrossRef]
- Haddad, N.; Brake, M.; de Miranda, J.R.; Migdadi, H. First detection of honey bee viruses in Jordan by RT-PCR. Jordanian J. Agric. 2008, 4, 242–247. [Google Scholar]
- Cornman, R.S.; Schatz, M.C.; Spencer, J.; Chen, Y.P.; Pettis, J.; Hunt, G.; Bourgeois, L.; Elsik, C.; Anderson, D.; Grozinger, C.M.; et al. Genomic survey of the ectoparasitic mite Varroa destructor, a major pest of the honey bee Apis mellifera. BMC Genomics 2010, 11, e602. [Google Scholar] [CrossRef] [PubMed]
- Cornman, R.S.; Tarpy, D.R.; Chen, Y.; Jeffreys, L.; Lopez, D.; Pettis, J.S.; vanEngelsdorp, D.; Evans, J.D. Pathogen webs in collapsing honeybee colonies. PLoS ONE 2012, 7, e43562. [Google Scholar] [CrossRef] [PubMed]
- Cornman, R.S.; Boncristiani, H.; Dainat, B.; Chen, Y.; vanEngelsdorp, D.; Weaver, D.; Evans, J.D. Population-genomic variation within RNA viruses of the Western honey bee, Apis mellifera, inferred from deep sequencing. BMC Genomics 2013, 14, e154. [Google Scholar] [CrossRef] [PubMed]
- Tentcheva, D.; Gauthier, L.; Bagny, L.; Fievet, J.; Dainat, B.; Cousserans, F.; Colin, M.E.; Bergoin, M. Comparative analysis of deformed wing virus (DWV) RNA in Apis mellifera L. and Varroa destructor. Apidologie 2006, 37, 41–50. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Snedecor, G.W.; Cochran, W.G. Statistical Methods, 7th ed.; The Iowa State University Press: Ames, IA, USA, 1980; pp. 1–507. [Google Scholar]
- Evans, J.D.; Schwarz, R.S.; Chen, Y.P.; Budge, G.; Cornman, R.S.; de La Rua, P.; de Miranda, J.R.; Foret, S.; Foster, L.; Gauthier, L.; et al. Standard methodologies for molecular research in Apis mellifera. In The COLOSS BEEBOOK, Volume I: Standard Methods for Apis Mellifera Research; Dietemann, V., Ellis, J.D., Neumann, P., Eds.; IBRA: Treforest, UK, 2013; Chapter 14; pp. 1–53. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.G.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, e539. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: New York, USA, 1965; pp. 97–166. [Google Scholar]
- Martelli, G.P.; Sabanadzovic, S.; Abou-Ghanem Sabanadzovic, N.; Edwards, M.C.; Dreher, T. The family Tymoviridae. Arch. Virol. 2002, 147, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.P.; Sabanadzovic, S.; Abou-Ghanem Sabanadzovic, N.; Saldarelli, P. (2002) Maculavirus, a new genus of plant viruses. Arch. Virol. 2002, 147, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S.; Tanaka, S.; Omuro, N.; Takabuchi, L.; Daimon, T.; Imanishi, S.; Yamashita, S.; Iwanaga, M.; Mita, K.; Maeda, S.; et al. Novel macula-like virus identified in Bombyx mori cultured cells. J. Virol. 2005, 79, 5577–5584. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lv, X.; Zhai, Y.; Fu, S.; Wang, D.; Rayner, S.; Tang, Q.; Liang, G. Genomic characterization of a novel virus of the family Tymoviridae isolated from mosquitoes. PLoS ONE 2012, 7, e39845. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, P.; Aumeier, P.; Ziegelmann, B. Biology and control of Varroa destructor. J. Invertebr. Pathol. 2010, 103, S96–S119. [Google Scholar] [CrossRef] [PubMed]
- Jakubiec, A.; Drugeon, G.; Camborde, L.; Jupin, I. Proteolytic processing of turnip yellow mosaic virus replication proteins and functional impact on infectivity. J. Virol. 2007, 81, 11402–11412. [Google Scholar] [CrossRef] [PubMed]
- Rozanov, M.N.; Drugeon, G.; Haenni, A.L. Papain-like proteinase of turnip yellow mosaic virus: A prototype of a new viral proteinase group. Arch. Virol. 1995, 140, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Lucas, W.J. Plant viral movement proteins: Agents for cell-to-cell trafficking of viral genomes. Virology 2006, 344, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, W.X.; Xie, D.; Peng, J.R.; Ding, S.W. Viral virulence protein suppresses RNA silencing-mediated defense but upregulates the role of microRNA in host gene expression. Plant Cell 2004, 16, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.W.; Howe, J.; Keese, P.; Mackenzie, A.; Meek, D.; Osorio-Keese, M.; Skotnicki, M.; Srifah, P.; Torronen, M.; Gibbs, A. The tymobox, a sequence shared by most tymoviruses: Its use in molecular studies of tymoviruses. Nucleic Acids Res. 1990, 18, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Alabdulla, A.; Minafra, A.; Elbeaino, T.; Saponari, M.; Savino, V.; Martelli, G.P. Complete nucleotide sequence and genome organization of Olive latent virus 3, a new putative member of the family Tymoviridae. Virus Res. 2010, 152, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.C.; Zhang, Z.; Weiland, J.J. Oat blue dwarf marafivirus resembles the tymoviruses in sequence, genome organization, and expression strategy. Virology 1997, 232, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Ravoet, J.; Maharramov, J.; Meeus, I.; de Smet, L.; Wenseleers, T.; Smagghe, G.; de Graaf, D.C. Comprehensive bee pathogen screening in Belgium reveals Crithidia mellificae as a new contributory factor to winter mortality. PLoS ONE 2013, 8, e72443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, B.; Forsgren, E.; Fries, I.; de Miranda, J.R. Acaricide treatment affects viral dynamics in Varroa destructor-infested honey bee colonies via both host physiology and mite control. Appl. Environ. Microbiol. 2012, 78, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Locke, B.; Forsgren, E.; de Miranda, J.R. Increased tolerance and resistance to virus infections: A possible factor in the survival of Varroa destructor-resistant honey bees (Apis mellifera). PLoS ONE 2014, 9, e99998. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Ravallec, M.; Tournaire, M.; Cousserans, F.; Bergoin, M.; Dainat, B.; de Miranda, J.R. Viruses associated with ovarian degeneration in Apis mellifera L. queens. PLoS ONE 2011, 6, e16217. [Google Scholar] [CrossRef] [PubMed]
- Harizanis, P.C. Infestation of queen cells by the mite Varroa jacobsoni. Apidologie 1991, 22, 533–538. [Google Scholar] [CrossRef]
- Williams, G.R.; Rogers, R.E.L.; Kalkstein, A.L.; Taylor, B.A.; Shutler, D.; Ostiguy, N.A. Deformed wing virus in western honey bees (Apis mellifera) from Atlantic Canada and the first description of an overtly-infected emerging queen. J. Invertebr. Pathol. 2009, 101, 77–79. [Google Scholar] [CrossRef] [PubMed]
- de Miranda, J.R.; Fries, I. Venereal and vertical transmission of deformed wing virus in honey bees (Apis mellifera L). J. Invertebr. Pathol. 2008, 98, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Schröder, M.; Gisder, S.; Genersch, E. Vertical-transmission routes for deformed wing virus of honeybees (Apis mellifera). J. Gen. Virol. 2007, 88, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Highfield, A.C.; Brettell, L.; Villalobos, E.M.; Budge, G.E.; Powell, M.; Nikaido, S.; Schroeder, D.C. Global honey bee viral landscape altered by a parasitic mite. Science 2012, 336, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Mondet, F.; de Miranda, J.R.; Kretzschmar, A.; le Conte, Y.; Mercer, A.R. On the front line: Quantitative virus dynamics in honeybee (Apis mellifera L.) colonies along a new expansion front of the parasite Varroa destructor. PLoS Pathog. 2014, 10, e1004323. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Miranda, J.R.; Cornman, R.S.; Evans, J.D.; Semberg, E.; Haddad, N.; Neumann, P.; Gauthier, L. Genome Characterization, Prevalence and Distribution of a Macula-Like Virus from Apis mellifera and Varroa destructor. Viruses 2015, 7, 3586-3602. https://0-doi-org.brum.beds.ac.uk/10.3390/v7072789
De Miranda JR, Cornman RS, Evans JD, Semberg E, Haddad N, Neumann P, Gauthier L. Genome Characterization, Prevalence and Distribution of a Macula-Like Virus from Apis mellifera and Varroa destructor. Viruses. 2015; 7(7):3586-3602. https://0-doi-org.brum.beds.ac.uk/10.3390/v7072789
Chicago/Turabian StyleDe Miranda, Joachim R., R. Scott Cornman, Jay D. Evans, Emilia Semberg, Nizar Haddad, Peter Neumann, and Laurent Gauthier. 2015. "Genome Characterization, Prevalence and Distribution of a Macula-Like Virus from Apis mellifera and Varroa destructor" Viruses 7, no. 7: 3586-3602. https://0-doi-org.brum.beds.ac.uk/10.3390/v7072789