
Therapeutics for Graft-versus-Host Disease: From Conventional Therapies to Novel Virotherapeutic Strategies



Abstract
:
1. Introduction
2. Clinical and Biological Overview of Graft-versus-Host Disease
2.1. Allogeneic Hematopoietic Stem Cell Transplantation Is the Only Potential Cure for Many Blood-Related Diseases, However the Practice Is Limited by Graft-versus-Host Disease
2.1.1. Sources of Hematopoietic Stem Cells Influence the Development of GVHD
2.1.2. Conditioning, Graft Manipulation and Post-Transplant Procedures Affect the Success of Allo-HSCT
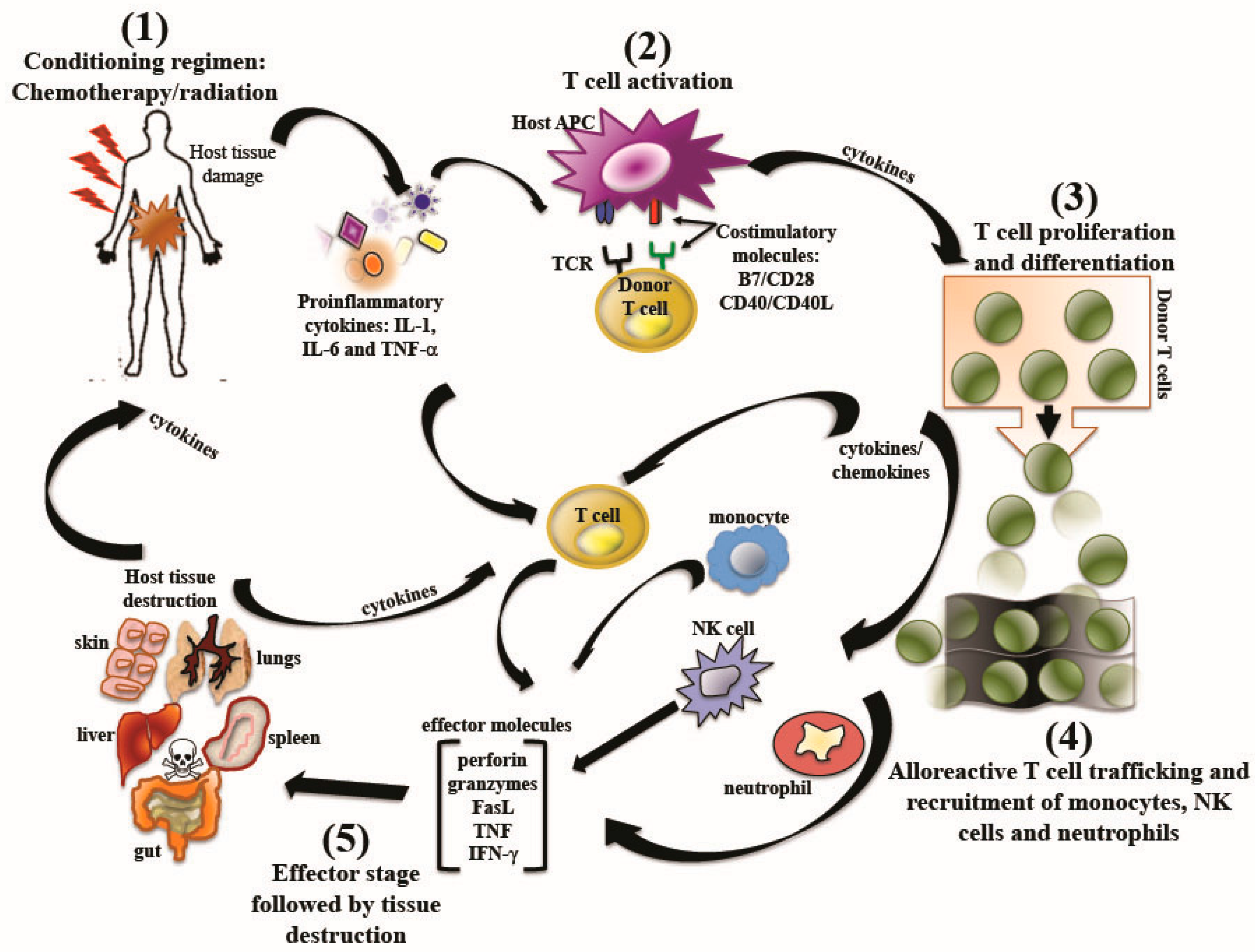
2.2. Graft-versus-Host Disease: Pathogenesis
2.3. Graft-versus-Host Disease: Clinical Presentations
2.3.1. Acute Graft-versus Host Disease
HLA Disparity
Conditioning Regimens
2.3.2. Chronic Graft-versus-Host Disease
3. Prevention of Graft-versus-Host Disease: Current Clinical Principles and Strategies
3.1. Limiting Organ Toxicity by Reducing Intensity of Chemotherapy Conditioning
3.2. Pharmacological Agents Used in Conventional Graft-versus-Host Therapies: Advantages and Drawbacks
3.2.1 Inhibition of Allo-Reactive T cells
Methotrexate (MTX)
Calcineurin Inhibitors
Corticosteroids
Cyclophosphamide
Mycophenolate Mofetil (MMF)
Sirolimus
3.2.2. Depletion of T lymphocytes
Monoclonal Antibodies (MAbs)
Positive selection of CD34+
4. Treatment of Established Graft-versus-Host-Disease
5. Alternative and Novel Strategies to Inhibit Graft-versus-Host Disease
5.1. Attenuating Graft-versus-Host Disease by B Cell Depletion
5.2. Cellular Suicide Gene Therapy
5.3. Regulatory T Cells
5.4. Targeting Co-Stimulatory Molecules
5.4.1. CD137
5.4.2. CD28
5.4.3. Inducible Co-Stimulator (ICOS)
5.4.4. Programmed Death 1 (PD-1)
5.4.5. The CD40/CD40-Ligand (CD40L) Pathway
5.5. Molecular Targets in T Cells That Modulate Graft-versus-Host Responses
5.5.1. Epigenetic Regulation in Donor T Cells
5.5.2. Targeting Notch Signals in Donor T Cells
5.5.3. Targeting Mitochondrial ATPase
5.5.4. Proteasome Inhibition
5.5.5. Targeting Protein Kinases
5.5.6. Therapeutic Intervention of GVHD by Targeting microRNAs
5.5.7. Targeting Janus Kinase/Signal Transducer and Activator of Transcription Pathways
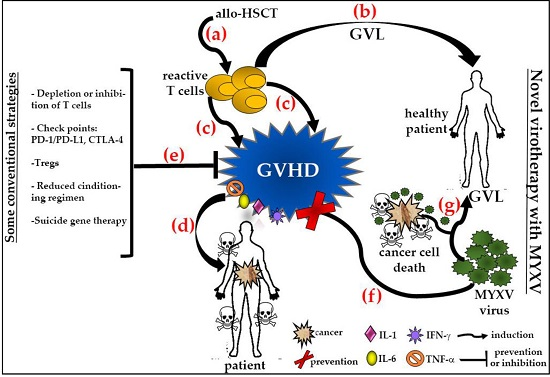
5.6. Virotherapy as a Novel Strategy to Prevent or Control Graft-versus-Host Disease and Augment the Graft-versus-Cancer Effects after Allogeneic Hematopoietic Stem Cell Transplantation
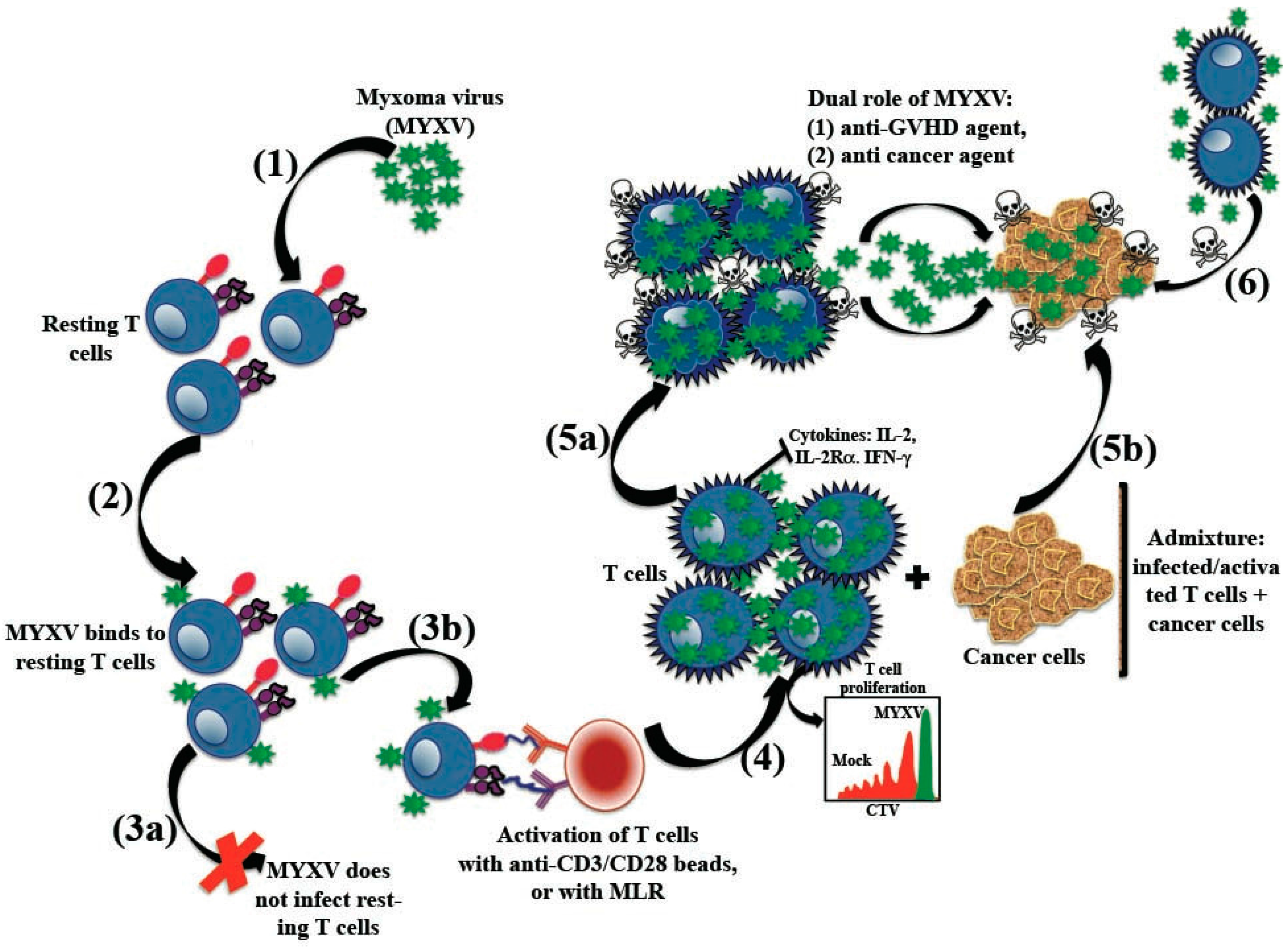
5.6.1. Ex Vivo Treatment with MYXV Prevents GVHD in a Xeno-Transplanted NSG Murine Model
5.6.2. MYXV Effects on Human T Cells
5.6.3. MYXV Has a Dual Role as Anti-GVHD and Anti-Cancer Agent
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Blazar, B.R.; Murphy, W.J.; Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 2012, 12, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Jamil, M.O.; Mineishi, S. State-of-the-art acute and chronic GVHD treatment. Int. J. Hematol. 2015, 101, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Weiden, P.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Feter, A.; Buckner, C.D.; Storb, R. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N. Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 273, 1550–1561. [Google Scholar] [CrossRef]
- Bais, S.; Bartee, E.; Rahman, M.M.; McFadden, G.; Cogle, C.R. Oncolytic virotherapy for hematological malignancies. Adv. Virol. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; Bartee, E.C.; Moreb, J.S.; Dower, K.; Connor, J.H.; McFadden, G. Myxoma and vaccinia viruses bind differentially to human leukocytes. J. Virol. 2013, 87, 4445–44460. [Google Scholar] [CrossRef] [PubMed]
- Villa, N.Y.; Bartee, E.; Mohamed, M.R.; Rahman, M.M.; Barrett, J.W.; McFadden, G. Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology 2010, 401, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; Chan, W.M.; Moreb, J.S.; Cogle, C.R.; McFadden, G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol. Blood Marrow Transplant. 2012, 18, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Madlambayan, G.J.; Bartee, E.; Kim, M.; Rahman, M.M.; Meacham, A.; Scott, E.W.; McFadden, G.; Cogle, C.R. Acute myeloid leukemia targeting by myxoma virus in vivo depends on cell binding but not permissiveness to infection in vitro. Leuk Res. 2012, 36, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; McFadden, G. Oncolytic Poxviruses. Annu. Rev. Virol. 2014, 1, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Rahman, M.M.; Cogle, C.R.; McFadden, G. Prevention of EBV lymphoma development by oncolytic myxoma virus in a murine xenograft model of post-transplant lymphoproliferative disease. Biochem. Buophys Res. Commun. 2015, 462, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; Meacham, A.; Wise, E.; Cogle, C.R.; McFadden, G. Virotherapy using myxoma virus prevents lethal graft-versus-host disease following xeno-transplantation with primary human hematopoietic stem cells. PLoS ONE 2012, 7, e43298. [Google Scholar] [CrossRef] [PubMed]
- Villa, N.Y.; Wasserfall, C.H.; Meacham, A.M.; Wise, E.; Chan, W.; Wingard, J.R.; McFadden, G.; Cogle, C.R. Myxoma virus suppresses proliferation of activated T lymphocytes yet permits oncolytic virus transfer to cancer cells. Blood 2015, 125, 3778–3788. [Google Scholar] [CrossRef] [PubMed]
- Baron, F.; Storb, R. Allogeneic hematopoietic cell transplantation as treatment for hematological malignancies: A review. Springer Semin. Immunopathol. 2004, 26, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L.O.; Simons, E.L.; Marrks, E.K.; Robson, M.J.; Bethard, W.F.; Gaston, E.O. The role of the spleen in radiation injury and recovery. J. Lab. Clin. Med. 1950, 35, 746–770. [Google Scholar] [PubMed]
- Lorenz, E.; Uphoff, D.; Reid, T.R.; Shelton, E. Modification of irradiation injury in mice and guinea pigs by bone marrow injections. J. Natl. 1951, 12, 197–201. [Google Scholar]
- Gatti, R.A.; Meuwissen, H.J.; Allen, H.D.; Hong, R.; Good, R.A. Immunological reconstitution of sexlinked lymphopenic immunological deficiency. Lancet 1968, 2, 1366–1369. [Google Scholar] [CrossRef]
- Rocha, V.; Locatelli, F. Searching for alternative hematopoietic stem cell donors for pediatric patients. Bone Marrow Transplant. 2008, 41, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, N.; Dreger, P.; Suttorp, M.; Rohwedder, E.B.; Haferlach, T.; Löffler, H.; Hunter, A.; Russell, N.H. Primary transplantation of allogeneic peripheral blood progenitor cells mobilized by filgrastim (granulocyte colony-stimulating factor). Blood 1995, 85, 1666–1672. [Google Scholar] [PubMed]
- Welniak, L.A.; Blazar, B.R.; Murphy, W.J. Immunobiology of allogeneic hematopoietic stem cell transplantation. Annu. Rev. Immunol. 2007, 25, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Barker, J.N.; Weisdorf, D.J.; DeFor, T.E.; Blazar, B.R.; McGlave, P.B.; Miller, J.S.; Verfaillie, C.M.; Wagner, J.E. Transplantation of 2 partially HLA-matched umbilical cord blood units to enhance engraftment in adults with hematologic malignancy. Blood 2005, 105, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.; Labopin, M.; Sanz, G.; Arcese, W.; Schwerdtfeger, R.; Bosi, A.; Jacobsen, N.; Ruutu, T.; de Lima, M.; Finke, J.; et al. Transplants of umbilical-cord blood or bone marrow from unrelated donors in adults with acute leukemia. N. Engl. J. Med. 2004, 351, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, M.J.; Eapen, M.; Rubinstein, P.; Wagner, J.E.; Zhang, M.J.; Champlin, R.E.; Stevens, C.; Barker, J.N.; Gale, R.P.; Lazarus, H.M.; et al. Outcomes after transplantation of cord blood or bone marrow from unrelated donors in adults with leukemia. N. Engl. J. Med. 2004, 351, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Holtick, U.; Chemnitz, J.M.; Hallek, M.; Scheid, C. Allogeneic haematopoietic stem cell transplantation—An overview. Klin. Monbl. Augenheiikd. 2015, 232, 641–646. [Google Scholar]
- Markey, K.A.; MacDonald, K.P.; Hill, G.R. The biology of graft-versus-host disease: Experimental systems instructing clinical practice. Blood 2014, 124, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Reddy, P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat. Rev. Clin. Oncol. 2014, 11, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, M.C.; Wang, Z.; Horowitz, M.M.; Gale, R.P. 2010 report from the Center for International Blood and Marrow Transplant Research (CIBMTR): Current uses and outcomes of hematopoietic cell transplants for blood and bone marrow disorders. Clin. Transpl. 2010, 87–105. [Google Scholar]
- Barnes, D.W.; Loutit, J.F.; Micklem, H.S. “Secondary disease” of radiation chimeras: A syndrome due to lymphoid aplasia. Ann. N. Y. Acad. Sci. 1962, 99, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Billingham, R.E. The biology of graft-versus-host reactions. Harvey Lect. 1977–1967, 62, 21–78. [Google Scholar]
- Korngold, R.; Sprent, J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T-cells from marrow. J. Exp. Med. 1978, 148, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, W.D.; Couzens, M.S.; Tang, C.B.; McNiff, J.; Robert, M.E.; Liu, J.; Shlomchik, M.J.; Emerson, S.G. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science 1999, 285, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Maeda, Y.; Liu, C.; Krijanovski, O.I.; Korngold, R.; Ferrara, J.L. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat. Med. 2005, 11, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Matte, C.C.; Liu, J.; Cormier, J.; Anderson, B.E.; Athanasiadis, I.; Jain, D.; McNiff, J.; Shlomchik, W.D. Donor APCs are required for maximal GVHD but not for GVL. Nat. Med. 2004, 10, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Paczesny, S.; Hanauer, D.; Sun, Y.; Reddy, P. New perspectives on the biology of acute GCHD. Bone Marrow Transplant. 2010, 45, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Hallek, M.J.; Storb, R.F.; von Bergwelt-Baildon, M.S. The role of B cells in the pathogenesis of graft-versus-host disease. Blood 2009, 114, 4919–4927. [Google Scholar] [CrossRef] [PubMed]
- Sarantopoulos, S.; Ritz, J. Aberrant B-cell homeostasis in chronic GVHD. Blood 2015, 125, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Won, J.H. B cell homeostasis and the development of chronic graft-versus-host disease: Implications for B cell-depleting therapy. Leuk. Lymphoma 2012, 53, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, C.A.; Panoskaltsis-Mortari, A.; Blazar, B.R.; Serody, J.S. Leukocyte migration and graft-versus-host disease. Blood 2005, 105, 4191–4199. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Penack, O.; Holler, E.; Idzko, M. Danger signals activating innate immunity in graft-versus-host disease. J. Mol. Med. (Berl.) 2011, 89, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.R.; Crawford, J.M.; Cooke, K.R.; Brinson, Y.S.; Pan, L.; Ferrara, J.L. Total body irradiation and acute graft-versus-host disease: The role of gastrointestinal damage and inflammatory cytokines. Blood 1997, 90, 3204–3213. [Google Scholar] [PubMed]
- Hill, G.R.; Ferrara, J.L.; Tabilio, A. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: Rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood 2000, 95, 2754–2759. [Google Scholar] [PubMed]
- Socié, G.; Blazar, B.R. Acute graft-versus-host disease: From the bench to the bedside. Blood 2009, 114, 4327–4336. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.Y.; Lowin, B.; French, L.; Acha-Orbea, H.; Tschopp, J. Cytotoxic T cells deficient in both functional fas ligand and perforin show residual cytolytic activity yet lose their capacity to induce lethal acute graft-versus-host disease. J. Exp. Med. 1996, 183, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Csencsits, K.L.; Bishop, D.K. Contrasting alloreactive CD4+ and CD8+ T cells: There's more to it than MHC restriction. Am. J. Transplant. 2003, 3, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Kagi, D.; Vignaux, F.; Ledermann, B.; Bürki, K.; Depraetere, V.; Nagata, S.; Hengartner, H.; Golstein, P. Fas and perforin pathways as major mechanisms of T cellmediated cytotoxicity. Science 1994, 265, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Van den Brink, M.R.; Burakoff, S.J. Cytolytic pathways in haematopoietic stem-cell transplantation. Nat. Rev. Immunol. 2002, 2, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Chicheportiche, Y.; Bourdon, P.R.; Xu, H.; Hsu, Y.M.; Scott, H.; Hession, C.; Garcia, I.; Browning, J.L. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 1997, 272, 32401–32410. [Google Scholar] [CrossRef] [PubMed]
- Schwab, L.; Goroncy, L.; Palaniyandi, S.; Gautam, S.; Triantafyllopoulou, A.; Mocsai, A.; Reichardt, W.; Karlsson, F.J.; Radhakrishnan, S.V.; Hanke, K.; et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat. Med. 2014, 20, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Giroux, M.; Delisle, J.S.; Gauthier, S.D.; Heinonen, K.M.; Hinsinger, J.; Houde, B.; Gaboury, L.; Brochu, S.; Wu, J.; Hébert, M.J.; et al. SMAD3 prevents graft-versus-host disease by restraining Th1 differentiation and granulocyte-mediated tissue damage. Blood 2011, 117, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Socié, G.; Mary, J.Y.; Lemann, M.; Daneshpouy, M.; Guardiola, P.; Meignin, V.; Ades, L.; Esperou, H.; Ribaud, P.; Devergie, A.; et al. Prognostic value of apoptotic cells and infiltrating neutrophils in graft-versus-host disease of the gastrointestinal tract in humans: TNF and Fas expression. Blood 2004, 103, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Tester, A.M.; Cox, J.H.; Connor, A.R.; Starr, A.E.; Dean, R.A.; Puente, X.S.; López-Otín, C.; Overall, C.M. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS ONE 2007, 2, e312. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.; Apperley, J.F.; Pavlů, J. Treatment and management of graft-versus-host disease: Improving response and survival. Ther. Adv. Hematol. 2013, 4, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Broady, R.; Yu, J.; Chow, V.; Tantiworawit, A.; Kang, C.; Berg, K.; Martinka, M.; Ghoreishi, M.; Dutz, J.; Levings, M.K. Cutaneous GVHD is associated with the expansion of tissue-localized Th1 and not Th17 cells. Blood 2010, 116, 5748–5751. [Google Scholar] [CrossRef] [PubMed]
- Imanguli, M.M.; Swaim, W.D.; League, S.C.; Gress, R.E.; Pavletic, S.Z.; Hakim, F.T. Increased T-bet+ cytotoxic effectors and type I interferon-mediated processes in chronic graft-versus-host disease of the oral mucosa. Blood 2009, 113, 3620–3630. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Welniak, L.A.; Taub, D.D.; Wiltrout, R.H.; Taylor, P.A.; Vallera, D.A.; Kopf, M.; Young, H.; Longo, D.L.; Blazar, B.R. Differential effects of the absence of interferon-gamma and IL-4 in acute graft-versus-host disease after allogeneic bone marrow transplantation in mice. J. Clin. Investig. 1998, 102, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, B.; Lee, S.; Bronson, R.T.; Grusby, M.J.; Sykes, M. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J. Clin. Investig. 2000, 105, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, P.; Janin, A.; Peffault de Latour, R.; Leboeuf, C.; Desveaux, A.; Keyvanfar, K.; Robin, M.; Clave, E.; Douay, C.; Quinquenel, A.; et al. Th17/Treg ratio in human graft-versus-host disease. Blood 2010, 116, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.E.; Inamoto, Y.; Carpenter, P.A.; Lee, S.J.; Kiem, H.P.; Petersdorf, E.W.; Pereira, S.E.; Nash, R.A.; Mielcarek, M.; Fero, M.L.; et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood 2011, 117, 3214–3219. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, J.L.; Reddy, P. Pathophysiology of graft-versus-host disease. Semin. Hematol. 2006, 43, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, J.L. Novel strategies for the treatment and diagnosis of graft-versus-host-disease. Best Preact. Res. Clin. Haematol. 2007, 20, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Díez, E.; García-Díez, A.; Marín, A.; Fernández-Herrera, J. Life-threatening graft-vs-host disease. Clin. Dermatol. 2005, 23, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Flomenberg, N.; Baxter-Lowe, L.A.; Confer, D.; Fernandez-Vina, M.; Filipovich, A.; Horowitz, M.; Hurley, C.; Kollman, C.; Anasetti, C.; Noreen, H.; et al. Impact of HLA class I and class II high-resolution matching on outcomes of unrelated donor bone marrow transplantation: HLA-C mismatching is associated with a strong adverse effect on transplantation outcome. Blood 2004, 104, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Carreras, E.; Jiménez, M.; Gómez-García, V.; de la Cámara, R.; Martín, C.; Martínez, F.; Iriondo, A.; Sanz, G.; Cañizo, C.; Cabrera, R.; et al. Donor age and degree of HLA matching have a major impact on the outcome of unrelated donor haematopoietic cell transplantation for chronic myeloid leukaemia. Bone Marrow Transplant. 2006, 37, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Deeg, H.J.; Antin, J.H. The clinical spectrum of acute graft-versus-host disease. Semin. Hematol. 2006, 43, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Christianson, G.J.; Yoshimura, Y.; Sproule, T.J.; Jung, N.; Joyce, S.; Roopenian, D.C. Immunodominance of H60 is caused by an abnormally high precursor T cell pool directed against its unique minor histocompatibility antigen peptide. Immunity 2002, 17, 593–603. [Google Scholar] [CrossRef]
- Bolaños-Meade, J.; Vogelsang, G.B. Acute graft-versus-host disease. Clin. Adv. Hematol. Oncol. 2004, 2, 672–682. [Google Scholar] [PubMed]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Natute 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, J.L.; Levy, R.; Chao, N.J. Pathophysiologic mechanisms of acute graft-vs.-host disease. Biol. Blood Marrow Transplant. 1999, 5, 347–356. [Google Scholar] [CrossRef]
- Chen, X.; Das, R.; Komorowski, R.; Beres, A.; Hessner, M.J.; Mihara, M.; Drobyski, W.R. Blockade of interleukin-6 signaling augments regulatory T-cell reconstitution and attenuates the severity of graft-versus-host disease. Blood 2009, 114, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.; Ganesan, J.; Müller, T.; Dürr, C.; Grimm, M.; Beilhack, A.; Krempl, C.D.; Sorichter, S.; Gerlach, U.V.; Jüttner, E.; et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 2010, 16, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Núñez, G. Orchestrating inflammasomes. Science 2012, 337, 1299–1300. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, D.; Ganesan, J.; Bscheider, M.; Stickel, N.; Weber, F.C.; Guarda, G.; Follo, M.; Pfeifer, D.; Tardivel, A.; Ludigs, K.; et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J. Exp. Med. 2013, 210, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.C.; Rothstein, D.M.; Sayegh, M.H. Costimulatory pathways in transplantation: Challenges and new developments. Immunol. Rev. 2009, 229, 271–293. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Kwon, B.S.; Panoskaltsis-Mortari, A.; Kwak, K.B.; Peschon, J.J.; Taylor, P.A. Ligation of 4–1BB (CDw137) regulates graft-versus-host disease, graft-versus-leukemia, and graft rejection in allogeneic bone marrow transplant recipients. J. Immunol. 2001, 166, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Levy, R.B.; Mak, T.W.; Panoskaltsis-Mortari, A.; Muta, H.; Jones, M.; Roskos, M.; Serody, J.S.; Yagita, H.; Podack, E.R.; et al. CD30/CD30 ligand (CD153) interaction regulates CD4+ T cell-mediated graft-versus-host disease. J. Immunol. 2004, 173, 2933–2941. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Sharpe, A.H.; Chen, A.I.; Panoskaltsis-Mortari, A.; Lees, C.; Akiba, H.; Yagita, H.; Killeen, N.; Taylor, P.A. Ligation of OX40 (CD134) regulates graft-versus-host disease (GVHD) and graft rejection in allogeneic bone marrow transplant recipients. Blood 2003, 101, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Carreno, B.M.; Panoskaltsis-Mortari, A.; Carter, L.; Iwai, Y.; Yagita, H.; Nishimura, H.; Taylor, P.A. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-gamma-dependent mechanism. J. Immunol. 2003, 171, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Ritz, J. Induction of tumor immunity following allogeneic stem cell transplantation. Adv. Immunol. 2006, 90, 133–73. [Google Scholar] [PubMed]
- Chen, B.J.; Cui, X.; Sempowski, G.D.; Liu, C.; Chao, N.J. Transfer of allogeneic CD62L-memory T cells without graft-versus-host disease. Blood 2004, 103, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Joe, G.; Hexner, E.; Zhu, J.; Emerson, S.G.; Haferlach, T. Host-reactive CD8+ memory stem cells in graftversus-host disease. Nat. Med. 2005, 11, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Joe, G.; Hexner, E.; Zhu, J.; Emerson, S.G. Alloreactive memory T cells are responsible for the persistence of graft-versus-host disease. J. Immunol. 2005, 174, 3051–3058. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Matte-Martone, C.; Li, H.; Anderson, B.E.; Venketesan, S.; Sheng Tan, H.; Jain, D.; McNiff, J.; Shlomchik, W.D. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood 2008, 111, 2476–2484. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.E.; McNiff, J.; Yan, J.; Doyle, H.; Mamula, M.; Shlomchik, M.J.; Shlomchik, W.D. Memory CD4+ T cells do not induce graft-versus-host disease. J. Clin. Investig. 2003, 112, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.H.; Siegel, R.M.; Harlan, D.M.; O’Shea, J.J. T cell-directed therapies: Lessons learned and future prospects. Nat. Immunol. 2007, 8, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Qi, J.; Wang, M.G.; Sykes, M. Donor-derived interferon gamma separates graft-versus-leukemia effects and graft-versus-host disease induced by donor CD8 T cells. Blood 2002, 99, 4207–4215. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Delmonte, J., Jr; Jalonen, C.K.; Ferrara, J.L. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces severity of experimental graft-versus-host disease. Blood 1995, 86, 4422–4429. [Google Scholar] [PubMed]
- Reddy, P.; Teshima, T.; Hildebrandt, G.; Williams, D.L.; Liu, C.; Cooke, K.R.; Ferrara, J.L. Pretreatment of donors with interleukin-18 attenuates acute graft-versus-host disease via STAT6 and preserves graft-versus-leukemia effects. Blood 2003, 101, 2877–2885. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.H.; Kurasawa, K.; Smith, R.; Eckhaus, M.A.; Gress, R.E. Donor CD4-enriched cells of Th2 cytokine phenotype regulate graft-versus-host disease without impairing allogeneic engraftment in sublethally irradiated mice. Blood 1994, 84, 3540–3549. [Google Scholar] [PubMed]
- Kappel, L.W.; Goldberg, G.L.; King, C.G.; Suh, D.Y.; Smith, O.M.; Ligh, C.; Holland, A.M.; Grubin, J.; Mark, N.M.; Liu, C.; et al. IL-17 contributes to CD4-mediated graft-versus-host disease. Blood 2009, 113, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Zhao, D.; Lin, C.L.; Zhang, C.; Chen, Y.; Todorov, I.; LeBon, T.; Kandeel, F.; Forman, S.; Zeng, D. Absence of donor Th17 leads to augmented Th1 differentiation and exacerbated acute graft-versus-host disease. Blood 2008, 112, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.L.; Trenado, A.; Vasey, D.; Klatzmann, D.; Salomon, B.L. CD4(+)CD25(+) immunoregulatory T Cells: New therapeutics for graft-versus-host disease. J. Exp. Med. 2002, 196, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Edinger, M.; Hoffmann, P.; Ermann, J.; Drago, K.; Fathman, C.G.; Strober, S.; Negrin, R.S. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat. Med. 2003, 9, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Coghill, J.M.; Carlson, M.J.; Moran, T.P.; Serody, J.S. The biology and therapeutic potential of natural regulatory T-cells in the bone marrow transplant setting. Leuk. Lymphoma 2008, 49, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, H.; Maeda, Y.; Teshima, T.; Sugiyama, H.; Kobayashi, K.; Yamasuji, Y.; Kadohisa, S.; Uryu, H.; Takeuchi, K.; Tanaka, T.; et al. Synthetic retinoid Am80 ameliorates chronic graft-versus-host disease by down-regulating Th1 and Th17. Blood 2012, 119, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, H.; Maeda, Y.; Tanimoto, M. Chronic graft-versus-host disease: Disease biology and novel therapeutic strategies. Acta Med. Okayama 2013, 67, 1–8. [Google Scholar] [PubMed]
- Wolff, D.; Gerbitz, A.; Ayuk, F.; Kiani, A.; Hildebrandt, G.C.; Vogelsang, G.B.; Elad, S.; Lawitschka, A.; Socie, G.; Pavletic, S.Z.; et al. Consensus conference on clinical practice in chronic graft-versus-host disease (GVHD): First-line and topical treatment of chronic GVHD. Biol. Blood Marrow Transplant. 2010, 16, 1611–1628. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Maeda, Y.; Kobayashi, K.; Nishimori, H.; Matsuoka, K.; Fujii, N.; Kondo, E.; Tanaka, T.; Chen, L.; Azuma, M.; et al. Programmed death-1 pathway in host tissues ameliorates Th17/Th1-mediated experimental chronic graft-versus-host disease. J. Immunol. 2014, 193, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Dhir, S.; Slatter, M.; Skinner, R. Recent advances in the management of graft-versus-host disease. Arch. Dis. Child. 2014, 99, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Ruutu, T.; Gratwohl, A.; de Witte, T.; Afanasyev, B.; Apperley, J.; Bacigalupo, A.; Dazzi, F.; Dreger, P.; Duarte, R.; Finke, J.; et al. Prophylaxis and treatment of GVHD: EBMT-ELN working group recommendations for a standardized practice. Bone Marrow Transplant. 2014, 49, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Alyea, E.P.; Kim, H.T.; Ho, V.; Cutler, C.; DeAngelo, D.J.; Stone, R.; Ritz, J.; Antin, J.H.; Soiffer, R.J. Impact of conditioning regimen intensity on outcome of allogeneic hematopoietic cell transplantation for advanced acute myelogenous leukemia and myelodysplastic syndrome. Biol. Blood Marrow Transplant. 2006, 12, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.L.; Sandmaier, B.M.; Storer, B.; Maris, M.B.; Sorror, M.L.; Maloney, D.G.; Chauncey, T.R.; Storb, R.; Deeg, H.J. Myeloablative vs nonmyeloablative allogeneic transplantation for patients with myelodysplastic syndrome or acute myelogenous leukemia with multilineage dysplasia: A retrospective analysis. Leukemia 2006, 20, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Storb, R.; Deeg, H.J.; Farewell, V.; Doney, K.; Appelbaum, F.; Beatty, P.; Bensinger, W.; Buckner, C.D.; Clift, R.; Hansen, J.; et al. Marrow transplantation for severe aplastic anemia: Methotrexate alone compared with a combination of methotrexate and cyclosporine for prevention of acute graft-versus-host disease. Blood 1986, 68, 119–125. [Google Scholar] [PubMed]
- Storb, R.; Thomas, E.D.; Buckner, C.D.; Clift, R.A.; Johnson, F.L.; Fefer, A.; Glucksberg, H.; Giblett, E.R.; Lerner, K.G.; Neiman, P. Allogeneic marrow grafting for treatment of aplastic anemia. Blood 1974, 43, 157–180. [Google Scholar] [PubMed]
- Powles, R.L.; Clink, H.M.; Spence, D.; Morgenstern, G.; Watson, J.G.; Selby, P.J.; Woods, M.; Barrett, A.; Jameson, B.; Sloane, J.; et al. Cyclosporin A to prevent graft-versus-host disease in man after allogeneic bone-marrow transplantation. Lancet 1980, 1, 327–329. [Google Scholar] [CrossRef]
- Fay, J.W.; Wingard, J.R.; Antin, J.H.; Collins, R.H.; Piñeiro, L.A.; Blazar, B.R.; Saral, R.; Bierer, B.E.; Przepiorka, D.; Fitzsimmons, W.E.; et al. K506 (Tacrolimus) monotherapy for prevention of graft-versus-host disease after histocompatible sibling allogenic bone marrow transplantation. Blood 1996, 87, 3514–3519. [Google Scholar] [PubMed]
- Chao, N.J.; Chen, B.J. Prophylaxis and treatment of acute graft-versus-host disease. Semin. Hematol. 2006, 43, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Quellmann, S.; Schwarzer, G.; Hübel, K.; Greb, A.; Engert, A.; Bohlius, J. Corticosteroids for preventing graft-versus-host disease after allogeneic myeloablative stem cell transplantation. Cochrane Database Syst. Rev. 2008, 16, CD004885. [Google Scholar]
- Kumar, S.; Chen, M.G.; Gastineau, D.A.; Gertz, M.A.; Inwards, D.J.; Lacy, M.Q.; Tefferi, A.; Harmsen, W.S.; Litzow, M.R. Prophylaxis of graft-versus-host disease with cyclosporine-prednisone is associated with increased risk of chronic graft-versus-host disease. Bone Marrow Transplant. 2001, 27, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Kanakry, C.G.; Tsai, H.L.; Bolaños-Meade, J.; Smith, B.D.; Gojo, I.; Kanakry, J.A.; Kasamon, Y.L.; Gladstone, D.E.; Matsui, W.; Borrello, I.; et al. Single-agent GVHD prophylaxis with posttransplantation cyclophosphamide after myeloablative, HLA-matched BMT for AML, ALL, and MDS. Blood 2014, 124, 3817–3827. [Google Scholar] [CrossRef] [PubMed]
- Koreth, J.; Antin, J.H. Current and future approaches for control of graft-versus-host disease. Expert Rev. Hematol. 2008, 1, 111. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.A.; Johnston, L.; Parker, P.; McCune, J.S.; Storer, B.; Slattery, J.T.; Furlong, T.; Anasetti, C.; Appelbaum, F.R.; Lloid, M.E.; et al. A phase I/II study of mycophenolate mofetil in combination with cyclosporine for prophylaxis of acute graft-versus-host disease after myeloablative conditioning and allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2005, 11, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.; Graef, T.; Tapprich, C.; Vaupel, M.; Steidl, U.; Germing, U.; Fenk, R.; Hinke, A.; Haas, R.; Kobbe, G. Cyclosporine A and mycophenolate mofetil vs cyclosporine A and methotrexate for graft-versus-host disease prophylaxis after stem cell transplantation from HLA-identical siblings. Bone Marrow Transplant. 2005, 35, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Reisner, Y.; Martelli, M.F. Bone marrow transplantation across HLA barriers by increasing the number of transplanted cells. Immunol. Today 1995, 16, 437–440. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Yanik, G. Acute graft versus host disease: Pathophysiology, risk factors, and prevention strategies. Clin. Adv. Hematol. Oncol. 2005, 3, 415–419. [Google Scholar] [PubMed]
- Reisner, Y.; Itzicovitch, L.; Meshorer, A.; Sharon, N. Hemopoietic stem cell transplantation using mouse bone marrow and spleen cells fractionated by lectins. Proc. Natl. Acad. Sci. USA 1978, 75, 2933–2936. [Google Scholar] [CrossRef] [PubMed]
- Reisner, Y.; Kapoor, N.; O’Reilly, R.J.; Good, R.A. Allogeneic bone marrow transplantation using stem cells fractionated by lectins: VI, in vitro analysis of human and monkey bone marrow cells fractionated by sheep red blood cells and soybean agglutinin. Lancet 1980, 2, 1320–1324. [Google Scholar] [CrossRef]
- Reisner, Y.; Kapoor, N.; Kirkpatrick, D.; Pollack, M.S.; Dupont, B.; Good, R.A.; O’Reilly, R.J. Transplantation for acute leukaemia with HLA-A and B nonidentical parental marrow cells fractionated with soybean agglutinin and sheep red blood cells. Lancet 1981, 2, 327–331. [Google Scholar] [CrossRef]
- Reisner, Y.; Kapoor, N.; Kirkpatrick, D.; Pollack, M.S.; Cunningham-Rundles, S.; Dupont, B.; Hodes, M.Z.; Good, R.A.; O’Reilly, R.J. Transplantation for severe combined immunodeficiency with HLA-A,B,D,DR incompatible parental marrow cells fractionated by soybean agglutinin and sheep red blood cells. Blood 1983, 61, 341–348. [Google Scholar] [PubMed]
- Kernan, N.A.; Bordignon, C.; Keever, C.A.; Cunningham, I.; Castro-Malaspina, H.; Collins, N.H.; Small, T.N.; Brochstein, J.; Emanuel, D.; Laver, J.; et al. Graft failures after T cell depleted marrow transplants for leukemia: Clinical and in vitro characteristics. Transplant. Proc. 1989, 19 (Suppl. S7), 29–32. [Google Scholar]
- Hanash, A.M.; Levy, R.B. Donor CD4+CD25+ T cells promote engraftment and tolerance following MHC-mismatched hematopoietic cell transplantation. Blood 2005, 105, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Aversa, F.; Tabilio, A.; Velardi, A.; Cunningham, I.; Terenzi, A.; Falzetti, F.; Ruggeri, L.; Barbabietola, G.; Aristei, C.; Latini, P.; et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N. Engl. J. Med. 1998, 339, 1186. [Google Scholar] [CrossRef] [PubMed]
- Bethge, W.A.; Faul, C.; Bornhäuser, M.; Stuhler, G.; Beelen, D.W.; Lang, P.; Stelljes, M.; Vogel, W.; Hägele, M.; Handgretinger, R.; et al. Haploidentical allogeneic hematopoietic cell transplantation in adults using CD3/CD19 depletion and reduced intensity conditioning: An update. Blood Cells Mol. Dis. 2008, 40, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bertaina, A.; Merli, P.; Rutella, S.; Pagliara, D.; Bernardo, M.E.; Masetti, R.; Pende, D.; Falco, M.; Handgretinger, R.; Moretta, F.; et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood 2014, 124, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.E.; Thompson, J.S.; Carter, S.L.; Kernan, N.A. Effect of graft-versus-host disease prophylaxis on 3-year disease-free survival in recipients of unrelated donor bone marrow (T-cell Depletion Trial): A multi-centre, randomised phase II-III trial. Lancet 2005, 366, 733–741. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Hale, G.; Waldmann, H. Alemtuzumab (Campath-1H) in allogeneic stem cell transplantation: Where do we go from here? Transplant. Proc. 2004, 36, 1225–1227. [Google Scholar] [CrossRef] [PubMed]
- Kanda, J.; Lopez, R.D.; Rizzieri, D.A. Alemtuzumab for the prevention and treatment of graft-versus-host disease. Int. J. Hematol. 2011, 93, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.J.; Rowley, S.D.; Anasetti, C.; Chauncey, T.R.; Gooley, T.; Petersdorf, E.W.; van Burik, J.A.; Flowers, M.E.; Storb, R.; Appelbaum, F.R.; et al. A phase I-II clinical trial to evaluate removal of CD4 cells and partial depletion of CD8 cells from donor marrow for HLA-mismatched unrelated recipients. Blood 1999, 94, 192–199. [Google Scholar]
- Ho, V.T.; Kim, H.T.; Li, S.; Hochberg, E.P.; Cutler, C.; Lee, S.J.; Fisher, D.C.; Milford, E.; Kao, G.; Daley, H.; et al. Partial CD8+ T-cell depletion of allogeneic peripheral blood stem cell transplantation is insufficient to prevent graft-versus-host disease. Bone Marrow Transplant. 2004, 34, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Labopin, M.; Aversa, F.; Rowe, J.M.; Bunjes, D.; Lewalle, P.; Nagler, A.; Di Bartolomeo, P.; Lacerda, J.F.; Lupo Stanghellini, M.T.; et al. A survey of fully haploidentical hematopoietic stem cell transplantation in adults with high-risk acute leukemia: A risk factor analysis of outcomes for patients in remission at transplantation. Blood 2008, 112, 3574–3581. [Google Scholar] [CrossRef] [PubMed]
- Tamari, R.; Chung, S.; Papadopoulos, E.B.; Jakubowski, A.A.; Hilden, P.; Devlin, S.M.; Goldberg, J.D.; Perales, M.A.; Ponce, D.M.; Sauter, C.S.; et al. CD34-Selected Hematopoietic Stem Cell Transplants Conditioned with Myeloablative Regimens and Antithymocyte Globulin for Advanced Myelodysplastic Syndrome: Limited Graft-versus-Host Disease without Increased Relapse. Biol. Blood Marrow Transplant. 2015, 21, 2106–2114. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Oliveira, G.; Stanghellini, M.T.; Vago, L.; Bondanza, A.; Peccatori, J.; Cieri, N.; Marktel, S.; Mastaglio, S.; Bordignon, C.; et al. Improving the safety of cell therapy with the TK-suicide gene. Front. Pharmacol. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.J.; Rizzo, J.D.; Wingard, J.R.; Ballen, K.; Curtin, P.T.; Cutler, C.; Litzow, M.R.; Nieto, Y.; Savani, B.N.; Schriber, J.R.; et al. First- and second-line systemic treatment of acute graft-versus-host disease: Recommendations of the American Society of Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2012, 18, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.M.; Cruickshank, S.; Rodell, T.C.; Gooley, T.; Schuening, F.; Rowley, S.; David, D.; Brunvand, M.; Berryman, B.; Abhyankar, S.; et al. A randomized, placebo-controlled trial of oral beclomethasone dipropionate as a prednisone-sparing therapy for gastrointestinal graft-versus-host disease. Blood 2007, 109, 4557–4563. [Google Scholar] [CrossRef] [PubMed]
- Noce, C.W.; Gomes, A.; Shcaira, V.; Corrêa, M.E.; Moreira, M.C.; Silva Júnior, A.; Gonçalves, L.S.; Garnica, M.; Maiolino, A.; Torres, S.R. Randomized double-blind clinical trial comparing clobetasol and dexamethasone for the topical treatment of symptomatic oral chronic graft-versus-host diseas. Biol. Blood Marrow Transplant. 2014, 20, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Baron, F.; Humblet-Baron, S.; Ehx, G.; Servais, S.; Hannon, M.; Belle, L.; Lechanteur, C.; Briquet, A.; Giet, O.; Baudoux, E.; et al. Thinking out of the box--new approaches to controlling GVHD. Curr. Hematol. Malig. Rep. 2014, 9, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Burchert, A.; Lengerke, C.; Verbeek, M.; Maas-Bauer, K.; Metzelder, S.K.; Spoerl, S.; Ditschkowski, M.; Ecsedi, M.; Sockel, K.; et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: A multicenter survey. Leukemia 2015, 29, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Spoerl, S.; Mathew, N.R.; Bscheider, M.; Schmitt-Graeff, A.; Chen, S.; Mueller, T.; Verbeek, M.; Fischer, J.; Otten, V.; Schmickl, M.; et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014, 123, 3832–3842. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Flowers, M.E. Recognizing and managing chronic graft-versus-host disease. Hematol. Am. Soc. Hematol. Educ. Program. 2008, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.C.; Chen, M.; Mirsoian, A.; Grossenbacher, S.K.; Tellez, J.; Ames, E.; Sun, K.; Jagdeo, J.; Blazar, B.R.; Murphy, W.J.; et al. Treatment of chronic graft-versus-host disease with bortezomib. Blood 2014, 124, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Mulder, A.; Heidt, S.; Vergunst, M.; Roelen, D.L.; Claas, F.H. Proteasome inhibition profoundly affects activated human B cells. Transplantation 2013, 95, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Neubert, K.; Meister, S.; Moser, K.; Weisel, F.; Maseda, D.; Amann, K.; Wiethe, C.; Winkler, T.H.; Kalden, J.R.; Manz, R.A.; et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008, 17, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Kharfan-Dabaja, M.A.; Cutler, C.S. Rituximab for prevention and treatment of graft-versus-host disease. Int. J. Hematol. 2011, 93, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Klein, J.P.; Barrett, A.J.; Ringden, O.; Antin, J.H.; Cahn, J.Y.; Carabasi, M.H.; Gale, R.P.; Giralt, S.; Hale, G.A.; et al. Severity of chronic graft-versus-host disease: Association with treatment-related mortality and relapse. Blood 2002, 100, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Khouri, I.F.; Saliba, R.M.; Giralt, S.A.; Lee, M.S.; Okoroji, G.J.; Hagemeister, F.B.; Korbling, M.; Younes, A.; Ippoliti, C.; Gajewski, J.L.; et al. Nonablative allogeneic hematopoietic transplantation as adoptive immunotherapy for indolent lymphoma: Low incidence of toxicity, acute graft-versus-host disease, and treatment-related mortality. Blood 2001, 98, 3593–3599. [Google Scholar] [CrossRef]
- Khouri, I.F.; McLaughlin, P.; Saliba, R.M.; Hosing, C.; Korbling, M.; Lee, M.S.; Medeiros, L.J.; Fayad, L.; Samaniego, F.; Alousi, A.; et al. Eight-year experience with allogeneic stem cell transplantation for relapsed follicular lymphoma after nonmyeloablative conditioning with fludarabine, cyclophosphamide, and rituximab. Blood 2008, 111, 5530–5536. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Cooper, N.; Del Poeta, G.; Stipa, E.; Laura Evangelista, M.; Abruzzese, E.; Amadori, S. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood 2008, 112, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Springer, C.J.; Niculescu-Duvaz, I. Prodrug-activating systems in suicide gene therapy. J. Clin. Investig. 2000, 105, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Bondanza, A.; Valtolina, V.; Magnani, Z.; Ponzoni, M.; Fleischhauer, K.; Bonyhadi, M.; Traversari, C.; Sanvito, F.; Toma, S.; Radrizzani, M.; et al. Suicide gene therapy of graft-versus-host disease induced by central memory human T lymphocytes. Blood 2006, 107, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Mailly, L.; Leboeuf, C.; Tiberghien, P.; Baumert, T.; Robinet, E. Genetically engineered T-cells expressing a ganciclovir-sensitive HSV-tk suicide gene for the prevention of GvHD. Curr. Opin. Investig. Drugs 2010, 11, 559–570. [Google Scholar] [PubMed]
- Lupo-Stanghellini, M.T.; Provasi, E.; Bondanza, A.; Ciceri, F.; Bordignon, C.; Bonini, C. Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum. Gene Ther. 2010, 21, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Di Stasi, A.; Tey, S.K.; Krance, R.A.; Martinez, C.; Leung, K.S.; Durett, A.G.; Wu, M.F.; Liu, H.; Leen, A.M.; et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood 2014, 123, 3895–3905. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Taylor, P.A.; Lees, C.J.; Blazar, B.R. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood 2002, 99, 3493–3499. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.A.; Panoskaltsis-Mortari, A.; Swedin, J.M.; Lucas, P.J.; Gress, R.E.; Levine, B.L.; June, C.H.; Serody, J.S.; Blazar, B.R. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood 2004, 104, 3804–3812. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.; Gorsse, N.; Romagnoli, P.; Hudrisier, D.; van Meerwijk, J.P. Induction of antigen-specific tolerance to bone marrow allografts with CD4+CD25+ T lymphocytes. Blood 2004, 103, 4216–4221. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Ermann, J.; Edinger, M.; Fathman, C.G.; Strober, S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J. Exp. Med. 2002, 196, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Brown, J.A.; Freeman, G.J.; Hafler, D.A. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 2001, 167, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.E.; Petrone, A.L.; Ponath, P.D. Differentiation and expansion of T cells with regulatory function from human peripheral lymphocytes by stimulation in the presence of TGF-{beta}. J. Immunol. 2005, 174, 14460–1455. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.E.; Alstad, A.N.; Merindol, N.; Crellin, N.K.; Amendola, M.; Bacchetta, R.; Naldini, L.; Roncarolo, M.G.; Soudeyns, H.; Levings, M.K. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol. Ther. 2008, 16, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, C.; Zeng, L.; Li, L.; Li, Z.; Xu, K. Engineered regulatory T cells prevent graft-versus-host disease while sparing the graft-versus-leukemia effect after bone marrow transplantation. Leuk. Res. 2010, 34, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B. Intervention with costimulatory pathways as a therapeutic approach for graft-versus-host disease. Exp. Mol. Med. 2010, 42, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Taylor, P.A.; Linsley, P.S.; Vallera, D.A. In vivo blockade of CD28/CTLA4: B7/BB1 interaction with CTLA4-Ig reduces lethal murine graft-versus-host disease across the major histocompatibility complex barrier in mice. Blood 1994, 83, 3815–3825. [Google Scholar] [PubMed]
- Hakim, F.T.; Cepeda, R.; Gray, G.S.; June, C.H.; Abe, R. Acute graft-versus-host reaction can be aborted by blockade of costimulatory molecules. J. Immunol. 1995, 155, 1757–1766. [Google Scholar] [PubMed]
- Yu, X.Z.; Martin, P.J.; Anasetti, C. Role of CD28 in acute graft-versus-host disease. Blood 1998, 92, 2963–2970. [Google Scholar] [PubMed]
- Sang, W.; Zhou, C.; Cheng, N.; Li, Z.; Zeng, L.; Xu, K. Control of mouse graft-versus-host disease following allogeneic bone marrow transplantation by blocking the CD28/B7 signaling pathway with lentiviral vector-mediated RNA interference. Immunol. Lett. 2011, 136, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.A.; Panoskaltsis-Mortari, A.; Freeman, G.J.; Sharpe, A.H.; Noelle, R.J.; Rudensky, A.Y.; Mak, T.W.; Serody, J.S.; Blazar, B.R. Targeting of inducible costimulator (ICOS) expressed on alloreactive T cells down-regulates graft-versus-host disease (GVHD) and facilitates engraftment of allogeneic bone marrow (BM). Blood 2005, 105, 3372–3380. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, V.M.; Eng, J.M.; Ramirez-Montagut, T.; Tjoe, K.H.; Muriglan, S.J.; Kochman, A.A.; Terwey, T.H.; Willis, L.M.; Schiro, R.; Heller, G.; et al. R. Absence of inducible costimulator on alloreactive T cells reduces graft versus host disease and induces Th2 deviation. Blood 2005, 106, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Aoyama, K.; Taylor, P.A.; Koehn, B.H.; Veenstra, R.G.; Panoskaltsis-Mortari, A.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Yagita, H.; et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood 2013, 122, 3062–3073. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, Y.; Fujino, M.; Wang, Q.; Kimura, H.; Azuma, M.; Kubo, M.; Abe, R.; Li, X.K. Involvement of the programmed death-1/programmed death-1 ligand pathway in CD4+CD25+ regulatory T-cell activity to suppress alloimmune responses. Transplantation 2007, 83, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Durie, F.H.; Aruffo, A.; Ledbetter, J.; Crassi, K.M.; Green, W.R.; Fast, L.D.; Noelle, R.J. Antibody to the ligand of CD40, gp39, blocks the occurrence of the acute and chronic forms of graft-vs-host disease. J. Clin. Investig. 1994, 94, 1333–1118. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Buhlman, J.; Xu, J.; Flavell, R.A.; Korngold, R.; Noelle, R.; Vallera, D.A. Blockade of CD40 ligand-CD40 interaction impairs CD4+ T cell-mediated alloreactivity by inhibiting mature donor T cell expansion and function after bone marrow transplantation. J. Immunol. 1997, 158, 29–39. [Google Scholar] [PubMed]
- Buhlmann, J.E.; Gonzalez, M.; Ginther, B.; Panoskaltsis-Mortari, A.; Blazar, B.R.; Greiner, D.L.; Rossini, A.A.; Flavell, R.; Noelle, R.J. Cutting edge: Sustained expansion of CD8+ T cells requires CD154 expression by Th cells in acute graft versus host disease. J. Immunol. 1999, 162, 4373–4376. [Google Scholar] [PubMed]
- Tamada, K.; Tamura, H.; Flies, D.; Fu, Y.X.; Celis, E.; Pease, L.R.; Blazar, B.R.; Chen, L. Blockade of LIGHT/LTbeta and CD40 signaling induces allospecific T cell anergy, preventing graft-versus-host disease. J. Clin. Investig. 2002, 109, 549–557. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.P.; Shlomchik, W.D.; Reddy, P. Biology of graft-versus-host responses: Recent insights. Biol. Blood Marrow Transplant. 2013, 19, S10–S14. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Reddy, P. HDAC inhibition and graft versus host disease. Mol. Med. 2011, 17, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Sun, Y.; Toubai, T.; Duran-Struuck, R.; Clouthier, S.G.; Weisiger, E.; Maeda, Y.; Tawara, I.; Krijanovski, O.; Gatza, E.; et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Investig. 2008, 118, 2562–2573. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, J.; Kato, K.; Xie, F.; Varambally, S.; Mineishi, S.; Kuick, R.; Mochizuki, K.; Liu, Y.; Nieves, E.; et al. Inhibition of histone methylation arrests ongoing graft-versus-host disease in mice by selectively inducing apoptosis of alloreactive effector T cells. Blood 2012, 119, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ritchey, J.; Prior, J.L.; Holt, M.; Shannon, W.D.; Deych, E.; Piwnica-Worms, D.R.; DiPersio, J.F. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood 2010, 116, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ebens, C.L.; Maillard, I. Notch signaling in hematopoietic cell transplantation and T cell alloimmunity. Blood Rev. 2013, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sandy, A.R.; Wang, J.R.V.; Shan, G.T.; Tran, I.T.; Friedman, A.; Kato, K.; He, S.; Cui, S.; Hexner, E.; et al. Notch signaling is a critical regulator of allogeneic CD4+ T-cell responses mediating graft-versus-host disease. Blood 2011, 117, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Gatza, E.; Wahl, D.R.; Opipari, A.W.; Sundberg, T.B.; Reddy, P.; Liu, C.; Glick, G.D.; Ferrara, J.L. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci. Transl. Med. 2011, 3, 67ra8. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Welniak, L.A.; Panoskaltsis-Mortari, A.; O’Shaughnessy, M.J.; Liu, H.; Barao, I.; Riordan, W.; Sitcheran, R.; Wysocki, C.; Serody, J.S.; et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc. Natl. Acad. Sci. USA 2004, 101, 8120–8125. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Wilkins, D.E.; Anver, M.R.; Sayers, T.J.; Panoskaltsis-Mortari, A.; Blazar, B.R.; Welniak, L.A.; Murphy, W.J. Differential effects of proteasome inhibition by bortezomib on murine acute graft-versus-host disease (GVHD): Delayed administration of bortezomib results in increased GVHD-dependent gastrointestinal toxicity. Blood 2005, 106, 3293–3299. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, J.O.; Iclozan, C.; Hossain, M.S.; Prlic, M.; Hopewell, E.; Bronk, C.C.; Wang, J.; Celis, E.; Engelman, R.W.; Blazar, B.R.; et al. PKCtheta is required for alloreactivity and GVHD but not for immune responses toward leukemia and infection in mice. J. Clin. Investig. 2009, 119, 3774–3786. [Google Scholar] [CrossRef] [PubMed]
- Berg-Brown, N.N.; Gronski, M.A.; Jones, R.G.; Elford, A.R.; Deenick, E.K.; Odermatt, B.; Littman, D.R.; Ohashi, P.S. PKCtheta signals activation versus tolerance in vivo. J. Exp. Med. 2004, 199, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.; Hermann-Kleiter, N.; Pfeifhofer-Obermair, C.; Lutz-Nicoladoni, C.; Thuille, N.; Letschka, T.; Barsig, J.; Baudler, M.; Li, J.; Metzler, B.; et al. PKC theta cooperates with PKC alpha in alloimmune responses of T cells in vivo. Mol. Immunol. 2009, 46, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Haarberg, K.M.; Li, J.; Heinrichs, J.; Wang, D.; Liu, C.; Bronk, C.C.; Kaosaard, K.; Owyang, A.M.; Holland, S.; Masuda, E.; et al. Pharmacologic inhibition of PKCα and PKCθ prevents GVHD while preserving GVL activity in mice. Blood 2013, 122, 2500–2511. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Heaphy, C.E.; Costinean, S.; Stauffer, N.; Na, C.; Hamadani, M.; Santhanam, R.; Mao, C.; Taylor, P.A.; Sandhu, S.; et al. Regulation of acute graft-versus-host disease by microRNA-155. Blood 2012, 119, 4786–4797. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.H.; Ziegler, J.; Li, C.; Sepulveda, A.; Bedeir, A.; Grandis, J.; Lentzsch, S.; Mapara, M.Y. Sequential activation of inflammatory signaling pathways during graft-versus-host disease (GVHD): Early role for STAT1 and STAT3. Cell Immunol. 2011, 268, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Lu, C.; Ziegler, J.; Liu, A.; Sepulveda, A.; Okada, H.; Lentzsch, S.; Mapara, M.Y. Absence of Stat1 in donor CD4⁺ T cells promotes the expansion of Tregs and reduces graft-versus-host disease in mice. J. Clin. Investig. 2011, 121, 2554–2569. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.; Amarnath, S.; Mariotti, J.; Kim, Y.C.; Foley, J.; Eckhaus, M.; O’Shea, J.J.; Fowler, D.H. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity 2012, 37, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Cetkovic-Cvrlje, M.; Roers, B.A.; Schonhoff, D.; Waurzyniak, B.; Liu, X.P.; Uckun, F.M. Treatment of post-bone marrow transplant acute graft-versus-host disease with a rationally designed JAK3 inhibitor. Leuk. Lymphoma 2002, 43, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Roers, B.A.; Waurzyniak, B.; Liu, X.P.; Cetkovic-Cvrlje, M. Janus kinase 3 inhibitor WHI-P131/JANEX-1 prevents graft-versus-host disease but spares the graft-versus-leukemia function of the bone marrow allografts in a murine bone marrow transplantation model. Blood 2002, 99, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.M.; McFadden, G. Myxoma virus and oncolytic virotherapy: A new biologic weapon in the war against cancer. Expert Opin. Biol. Ther. 2007, 7, 1415–1225. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Kerr, P.J. Myxomatosis in Australia and Europe: A model for emerging infectious diseases. Antivir. Res. 2012, 93, 387–415. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.M.; Werden, S.J.; McFadden, G. Myxoma virus in the European rabbit: Interactions between the virus and its susceptible host. Vet. Res. 2007, 38, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F. Adventures with poxviruses of vertebrates. FEMS Microbiol. Rev. 2000, 24, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Villa, N.Y.; Bais, S.; Chan, W.M.; Meacham, A.M.; Wise, E.; Rahman, M.M.; Moreb, J.S.; Rosenau, E.H.; Wingard, J.R.; McFadden, G.; et al. Ex vivo virotherapy with myxoma virus does not impair hematopoietic stem and progenitor cells. Cytotherapy 2016, 18, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; McFadden, G. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-beta. Cytokine 2009, 47, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; Mohamed, M.R.; Lopez, M.C.; Baker, H.V.; McFadden, G. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J. Virol. 2009, 83, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Barrett, J.W.; Stanford, M.; Werden, S.J.; Johnston, J.B.; Gao, X.; Sun, M.; Cheng, J.Q.; McFadden, G. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. USA 2006, 103, 4640–4645. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; McFadden, G. Pharmacological manipulation of the akt signaling pathway regulates myxoma virus replication and tropism in human cancer cells. J. Virol. 2010, 84, 3287–3302. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Liu, J.; Chan, W.M.; Rothenburg, S.; McFadden, G. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 2013, 9, e1003465. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; McFadden, G. The role of cell signaling in poxvirus tropism: The case of the M-T5 host range protein of myxoma virus. Biochim. Biophys. Acta 2008, 1784, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Williamson, C.T.; Prudhomme, J.; Bebb, D.G.; Riabowol, K.; Lee, P.W.; Lees-Miller, S.P.; Mori, Y.; Rahman, M.M.; McFadden, G.; et al. The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene 2010, 29, 3990–3996. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Madlambayan, G.J.; Cogle, C.R.; McFadden, G. Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with Myxoma virus. Cytokine Growth Factor Rev. 2010, 21, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Madlambayan, G.J.; Rahman, M.M.; Smallwood, S.E.; Meacham, A.M.; Hosaka, K.; Scott, E.W.; Cogle, C.R.; McFadden, G. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia 2009, 23, 2313–2317. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; Rahman, M.M.; McFadden, G. Oncolytic myxoma virus: The path to clinic. Vaccine 2013, 31, 4252–4258. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Mediators of aGVHD | Examples | Functions in the Context of aGVHD | Ref. |
|---|---|---|---|
| T cell co-receptors for MHC class I and class II | CD4+ (MHC-class II) and CD8+ (MHC class I) T cells. |
| [81] |
| Naïve and memory T cells | CD62L+ CD44− (naïve T cells); CD62L+ CD44+ (central memory T cells); CD62L− CD44− (terminally differentiated effector/effector memory). |
| [82,83,84,85,86] |
| Th1 subset | IFN-γ, IL-2 and TNF-α. |
| [87,88] |
| Th2 subset | IL-4, and IL-10, G-CSF, IL-4 and IL-18. |
| [34,59,89,90,91] |
| Th17 subset | IL-17 |
| [34,92,93] |
| Regulatory T cells (Tregs) | CD4+CD25+Foxp3+ (nTregs) |
| [94,95,96] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa, N.Y.; Rahman, M.M.; McFadden, G.; Cogle, C.R. Therapeutics for Graft-versus-Host Disease: From Conventional Therapies to Novel Virotherapeutic Strategies. Viruses 2016, 8, 85. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030085
Villa NY, Rahman MM, McFadden G, Cogle CR. Therapeutics for Graft-versus-Host Disease: From Conventional Therapies to Novel Virotherapeutic Strategies. Viruses. 2016; 8(3):85. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030085
Chicago/Turabian StyleVilla, Nancy Y., Masmudur M. Rahman, Grant McFadden, and Christopher R. Cogle. 2016. "Therapeutics for Graft-versus-Host Disease: From Conventional Therapies to Novel Virotherapeutic Strategies" Viruses 8, no. 3: 85. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030085