1. Introduction
Eurycomanone is uniquely found in
Eurycoma longifolia Jack (family Simaroubaceae), which is a herbaceous tree found mainly in Southeast Asia. Eurycomanone is reported to be the most abundant phytochemical quassinoid in
E. longifolia roots [
1,
2,
3,
4,
5,
6,
7,
8]. The MS: 2409:201 of producing a freeze-dried standardised water extract of
E. longifolia uses eurycomanone as the chemical marker. The eurycomanone level must be consistently present at 0.80–1.50%
w/
v, alongside other markers such as total polysaccharides, total protein, and total glycosaponin, where their levels are expected at >20%. Traditionally,
E. longifolia was used for ailments such as fever, wounds, and ulcers, as well as an afterbirth remedy or as a general tonic [
9,
10]. Biological activities of
E. longifolia previously reported, such as male fertility enhancement [
4,
11] and antimalarial [
12,
13,
14,
15,
16,
17], cytotoxic [
14,
15,
18], antiproliferative [
19,
20], and antiulcer [
21] effects, are largely attributed to the quassinoids group, specifically eurycomanone.
Studies identifying eurycomanone as the compound responsible for these reported activities mainly focused on in vitro systems. Previous investigation into the physicochemical properties of several quassinoids of
E. longifolia [
16] revealed that eurycomanone and 13-
α-(21)-epoxyeurycomanone possessed the necessary characteristics contributing to
E. longifolia’s effect. Favourable physicochemical properties such as solubility, lipophilicity, chemical stability, permeability, plasma stability, and plasma protein binding act as indicators of the marker’s behavior in the body. The behaviour of eurycomanone in the body is of interest as the consumption of
E. longifolia in the forms of water extracts incorporated into health supplements and beverages are the common strategies for marketed
E. longifolia-based products. Oral consumption of
E. longifolia in these various forms exposes its active ingredients to varying environment, which may affect their bioavailability in the body.
Bioavailability measures the delivery of an active ingredient to its site of action in order to cause the predicted effect(s). First-pass metabolism in the liver can markedly reduce the amount of eurycomanone available to the site of action, which can be predicted in vitro by assessing its metabolic stability prior to its administration orally. Previously reported eurycomanone bioavailability in rats was studied using extracts with differing amounts of eurycomanone [
7,
22,
23].
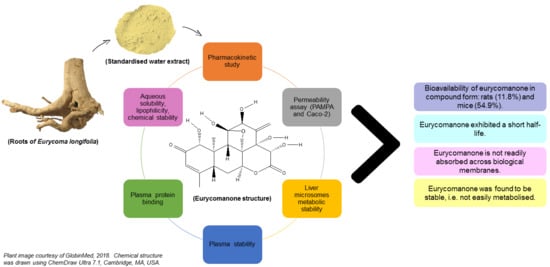
Experimentally based data on eurycomanone’s bioavailability will be highly essential if a standardised extract is to be clinically tested. Nevertheless, to date, no bioavailability study on the standardised water extract form has been previously conducted. This study investigates the bioavailability of eurycomanone in its pure form and in a standardised water extract of E. longifolia.
2. Materials and Methods
2.1. Chemicals and Reagents
Standardised water extract of E. longifolia used conformed to the MS: 2409:201 and contained eurycomanone (1.36%), total protein (30.5%), total polysaccharide (37.8%), and glycosaponin (52%). Eurycomanone (94.8% purity) was obtained from ChromaDex Inc. (Irvine, CA, USA). Propranolol, estriol, metoprolol tartrate, diethylstil bestrol, erythromycin, atenolol, carbamazepine, propantheline bromide, enalapril, dasatinib, midazolam, terfinadine, alamethicin, uridine 5′-diphospho-glucuronosyltransferase (UDPGA), nicotinamide adenine dinucleotide phosphate (NADPH), N-methyl-2-pyrrolidone (NMP), 2-hydroxypropyl-beta-cyclodextrin (HPCD), and chemicals used in reagents and buffer preparations were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Plasma and microsomes from mice and rats were prepared in-house, whilst plasma and microsomes from dog, monkey, and human were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Solvents such as methanol, formic acid, acetonitrile (ACN), and DMSO used were of LC-MS and HPLC grade and were purchased from Fisher Chemicals (Waltham, MA, USA).
2.2. Animal Experiments
2.2.1. Eurycomanone Assessment
The ethical approval for the animal experiments conducted in India was granted from the Animal Ethics Committee, Aurigene Discovery Technologies, Bangalore, India. Eight male Wistar rats (12 weeks old; weighing 250–350 g) and eight Caeserean Derived-1 (CD-1) mice (weighing 20–30 g) were obtained from the Animal House, Aurigene Discovery Technologies, Bangalore, India. They were supplied with standard rodent diets and drinking water ad libitum. Each rodent species (i.e., rats and mice) were divided into two groups. One group (n = 3 for each rodent species) was administered with eurycomanone via the intravenous route (IV) and another group (n = 3 for each rodent species) via the oral route (PO) using oral gavage intubation needle. Two animals of each of the species were used as a control and they received reverse osmosis water. All rats underwent jugular vein cannulation surgery 72 h prior to the administration of the eurycomanone.
2.2.2. Standardised Water Extract of E. longifolia (SWE) Assessment
The animal experiment conducted in Malaysia was approved by the Animal Care and Use Committee, Ministry of Health Malaysia (ACUC Number: ACUC/KKM/02(1/2014)). Ten male Sprague Dawley rats (12 weeks old; weighing 250–400 g) were obtained from the Laboratory Animal Resource Unit, Institute for Medical Research, Kuala Lumpur, Malaysia. They were supplied with standard rodent diets and drinking water ad libitum. One group (n = 4) was administered with SWE via the IV route and another group (n = 4) via the PO route using oral gavage intubation needle. Two rats were used as the control and they were administered reverse osmosis water. All rats underwent jugular vein cannulation surgery 72 h prior to the administration of SWE. All animals were maintained in a 12-h light and dark cycle, the temperature was maintained between 22 ± 3 °C, and the relative humidity between 50–65%.
2.3. Sample Preparation
Eurycomanone (rats: 1.5 mg/mL for IV route and 3.0 mg/mL for PO route; mice: 0.5 mg/mL for IV route and 1.0 mg/mL for PO route) samples were prepared fresh on the day of dosing. For the IV administration, eurycomanone was dissolved in 3% NMP and 97% of 10% HPCD in saline. For the oral administration, eurycomanone was dissolved in 3% NMP and 97% of 30% HPCD in saline. Both samples were sonicated before use. The SWE (5 mg/mL for IV route, 10 mg/mL for PO route) was prepared fresh on the day of dosing. For the IV administration, the SWE was dissolved in reverse osmosis water and filtered (0.2 µm pore size) before administration. The SWE for the PO administration was prepared using the same method, except without filtering. The animals were not fasted and doses given were based on the body weights prior to dosing.
2.4. Specimen Collection
Blood specimens (approximately 0.30–0.40 mL via the jugular vein cannula for rats and 0.10–0.20 mL via submanibular bleeding for mice) were collected at time points 0.083 (IV group only), 0.25, 0.5, 1, 2, 4, 6, and 8 h post-dosing with eurycomanone. At the experiment end point, the animals were euthanized by excess isoflurane inhalation followed by cervical dislocation. In the experiment using SWE, the rats were placed in metabolic cages (Techniplast, West Chester, PA, USA) and blood (approximately 0.30–0.40 mL collected via the jugular vein cannula), urine, and faeces specimens were collected at the same time points with the above plus one additional time point of 24 h. Kidney and liver specimens were collected at necropsy, where the rats were euthanized by CO2 inhalation followed by cervical dislocation. All blood specimens were centrifuged (10,000 rpm, 10 min) to obtain plasma and were stored at −80 °C until processed. Urine and faeces specimens were collected into plastic containers on wet ice while the organ specimens were snap-frozen in liquid nitrogen and maintained at −80 °C until processed.
2.5. Specimen Preparation for LC-MS/MS Analysis
Plasma specimens (100 µL), collected from the eurycomanone experiment were added with the internal standard solution (10 µL). Plasma proteins were precipitated with ACN (300 µL), vortex-mixed for 5 min, and centrifuged at 4000 rpm for 7 min. The supernatant was collected and evaporated using a nitrogen evaporator for 20 min at 50 °C. Thereafter, they were reconstituted with the mobile phase (500 µL) and transferred into LC-MS vials for analysis.
Plasma specimens (200 µL) from the SWE experiment were added with the internal standard solution (50 µL). Plasma proteins were precipitated with ACN (400 µL), vortex-mixed for 5 min, and centrifuged at 4000 rpm for 7 min. The supernatant was collected and evaporated to dryness using nitrogen evaporator at 60 °C. Thereafter, they were reconstituted with 80 µL of 30% methanol: 70% (0.1% formic acid) and transferred into LC-MS vials for analysis.
Urine specimens (200 µL) were added with ACN (400 µL), vortex-mixed for 5 min, and centrifuged at 4000 rpm for 7 min. The supernatant was collected and evaporated to dryness using a nitrogen evaporator at 60 °C. Thereafter, they were reconstituted with 80 µL of 10% methanol: 90% (0.1% formic acid) and transferred into LC-MS vials for analysis.
Faeces specimens were weighed, ultrapure water (at 8 times the weighed samples) was added, and samples were homogenised. The homogenised faeces samples (200 µL) were added with ACN (400 µL), vortex-mixed for 5 min, and centrifuged at 4000 rpm for 7 min. The supernatant was collected and evaporated to dryness using a nitrogen evaporator at 60 °C. Thereafter, they were reconstituted with 80 µL of 5% methanol: 95% (0.1% formic acid) and transferred into LC-MS vials for analysis.
Tissue specimens (kidneys and livers) were blotted with filter paper, weighed, and minced. Ultrapure water (at 3 times the weighed tissue samples) were added and samples were homogenised. The homogenised tissue specimens (200 µL) were added with ACN (400 µL), vortex-mixed for 5 min, and centrifuged at 4000 rpm for 7 min. The supernatant was collected and evaporated to dryness using a nitrogen evaporator at 60 °C. Thereafter, they were reconstituted with 80 µL of 5% methanol: 95% (0.1% formic acid) and transferred into LC-MS vials for analysis.
2.6. LC-MS/MS Analysis
Eurycomanone detection in the pure eurycomanone experiments was done via the LC-MS/MS system consisting of an Agilent 1260 Infinity HPLC system and an AB SCIEX API 4000™ Triple-Quadrupole mass spectrometer equipped with TurboIonSpray® probe and atmospheric pressure chemical ionization (APCI) (AB Sciex LLC, Framingham, MA, USA). Electrospray ionization (ESI) was performed in positive ion mode. The analytical column was Agilent Zorbax Eclipse-C18 (4.6 mm I.D. × 150 mm, 3.5 µm). The mobile phase consisted of 0.1% formic acid (solvent A) and 90% ACN with 0.1% formic acid (solvent B). The flow rate was set at 1.2 mL/min and the injection volume was 50 µL with the run time of 3.5 min. The mass transitions, monitored using multiple-reaction monitoring (MRM) detections, were 409 → 221.1/143.1 for eurycomanone. Eurycomanone detection in the SWE experiments were done via an LC-MS system consisting of Agilent 1100 series HPLC and mass spectrometer detector system with APCI-ESI ionization mode (Agilent Technologies, Santa Clara, CA, USA).
For plasma specimens, the analytical column used was Agilent Zorbax Eclipse XDB-Phenyl (2.1 mm I.D. × 50 mm, 5 µm). The mobile phase consisted of 35% methanol: 65% (0.1% formic acid). The flow rate was set at 0.2 mL/min and the injection volume was 2 µL with the run time of 5 min. For urine samples, the analytical column was Agilent Zorbax Eclipse XDB-Phenyl (2.1 mm I.D. × 50 mm, 3.5 µm). The mobile phase consisted of 10% methanol: 90% (0.1% formic acid). The flow rate was set at 0.2 mL/min and the injection volume was 2 µL with the run time of 5 min.
For faeces, kidney, and liver specimens, the analytical column used was Agilent Zorbax Eclipse XDB-Phenyl (2.1 mm I.D. × 50 mm, 3.5 µm). The mobile phase consisted of 5% methanol: 95% (0.1% formic acid). The flow rate was set at 0.2 mL/min and the injection volume was 2 µL with the run time of 6 min. The identification of eurycomanone and internal standard for plasma matrix were achieved by comparing the ion sets in selected ion monitoring (SIM) for eurycomanone with sodium adduct (m/z 431.0) and metronidazole (172.0 g/mol) in samples with the standard solution, at a similar retention time using LC-MS analysis. The concentration of the specimens analysed were determined by using an inverse prediction method for the best-fit regression curve using the weighted least-square (WLS) of , obtained from the standard concentrations.
In the analytical run, each standard in the calibration curve for each matrix was checked for the accuracy to be in the range of 80–120% for the lower limit of quantification (LLOQ) concentration and 85–115% for the remaining standards. Each quality control (QC) concentration was also checked for the accuracy to be in the range of 80–120% for the LLOQ concentration and 85–115% for the remaining QC samples. Sample matrices from the control group (blank samples) were used in the preparation of the calibration curve for determining eurycomanone concentrations. The pharmacokinetic analysis was performed using the non-compartmental analysis of PhoenixTM WinNolin® software (version 1.3, Certara, L.P., Princeton, NJ, USA).
2.7. Aqueous Solubility, Lipophilicity, and Chemical Stability of Eurycomanone
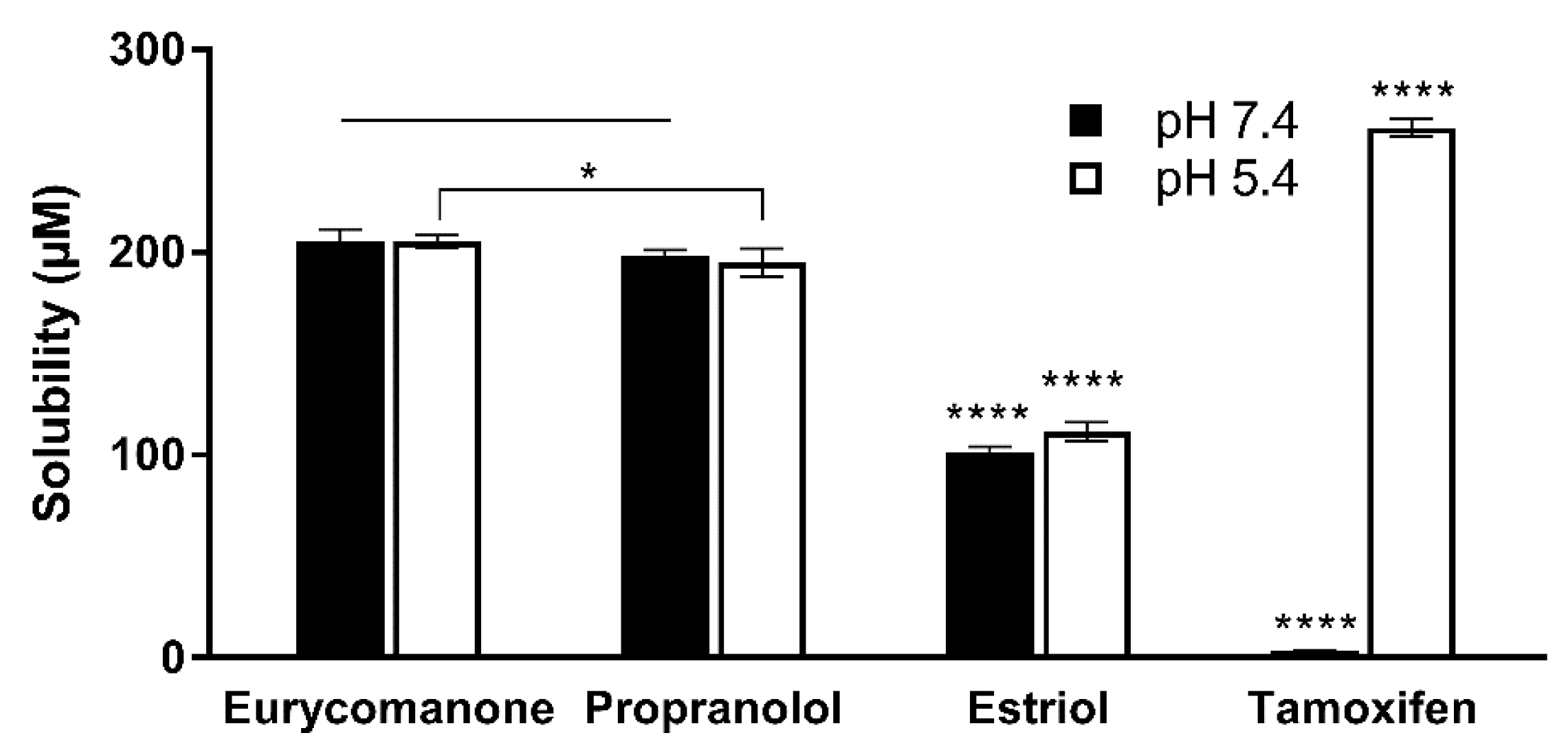
The aqueous stability was estimated by adding 10 µL of eurycomanone stock solution (10 mM) to 490 µL of aqueous buffer at pH 5.4, pH 7.4 and DMSO in triplicates in deep-well plates. Three standards (propanolol, estriol, and tamoxifen) at the same concentration were also added in the deep-well plates. The plate was kept on a shaker (300 rpm) at room temperature for 16 h. The plate was then centrifuged at 25 °C, 4000 rpm for 25 min. The supernatant was transferred and analysed using LC-MS/MS. The aqueous solubility was calculated using the following formula:
Peak area 1 = peak area of the compound in 2% DMSO at the different pH levels.
Peak area 2 = peak area of the compound in 100% DMSO.
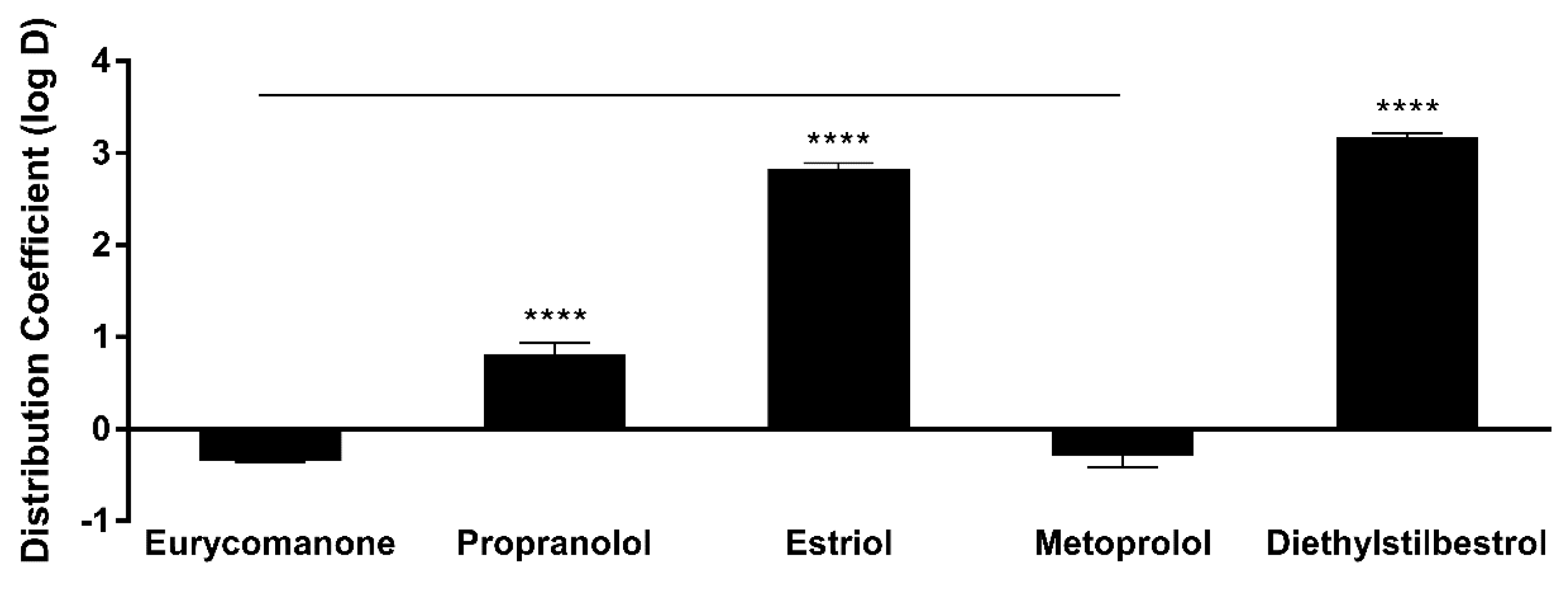
The lipophilicity of eurycomanone was estimated using the shake flask method with octanol and buffer (pH 7.4). Eurycomanone (5 µL) at 5 mM concentration is added to octanol (495 µL) and DMSO (495 µL) and also into the aqueous and octanol phase in a 1:1 ratio (247.4 µL of octanol phase and 247.5 µL of aqueous phase). The octanol and aqueous phases were prepared using a saturation process, where equal amounts of D-PBS (pH 7.4) and octanol were saturated (300 rpm, room temperature) for 24 h before being separated and kept at room temperature. The prepared mixtures were vortexed and kept on the shaker (300 rpm) at room temperature for 16 h. The mixtures were allowed to stand for 30 min, and 200 µL of solution was transferred and sent for HPLC analysis. The analyses were conducted along with propranolol, estriol, metoprolol tartrate, and diethylstil bestrol (5 mM) as standards and were conducted in triplicates.
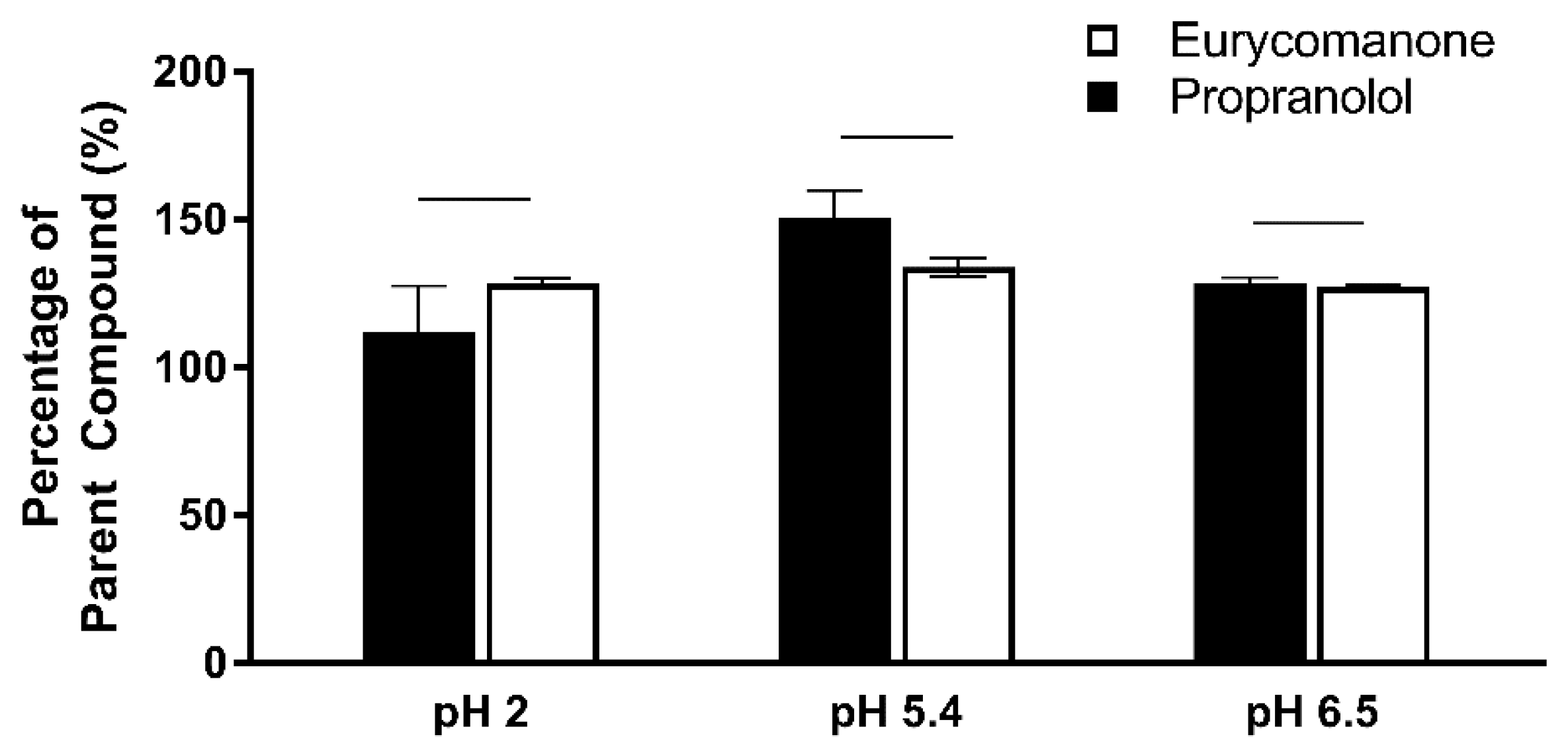
The chemical stability of eurycomanone was estimated in gastric simulated fluid (GSF), pH 2; fasted simulated intestinal fluid (FASSIF), pH 6.5; fed simulated intestinal fluid (FESSIF), pH 5.0; and in 150 mM NaHCO3 at pH 9.2. Eurycomanone (3 µL) at 5 mM was added to 297 µL of the respective buffers. The mixture (100 µL) was added to 100 µL of ACN at time points 1 and 3 h and sent for LC-MS/MS analysis. The test was done in triplicates with propranolol and erythromycin (5 mM) as standards.
2.8. Permeability Assay
2.8.1. Parallel Artificial Membrane Permeability Assay (PAMPA)
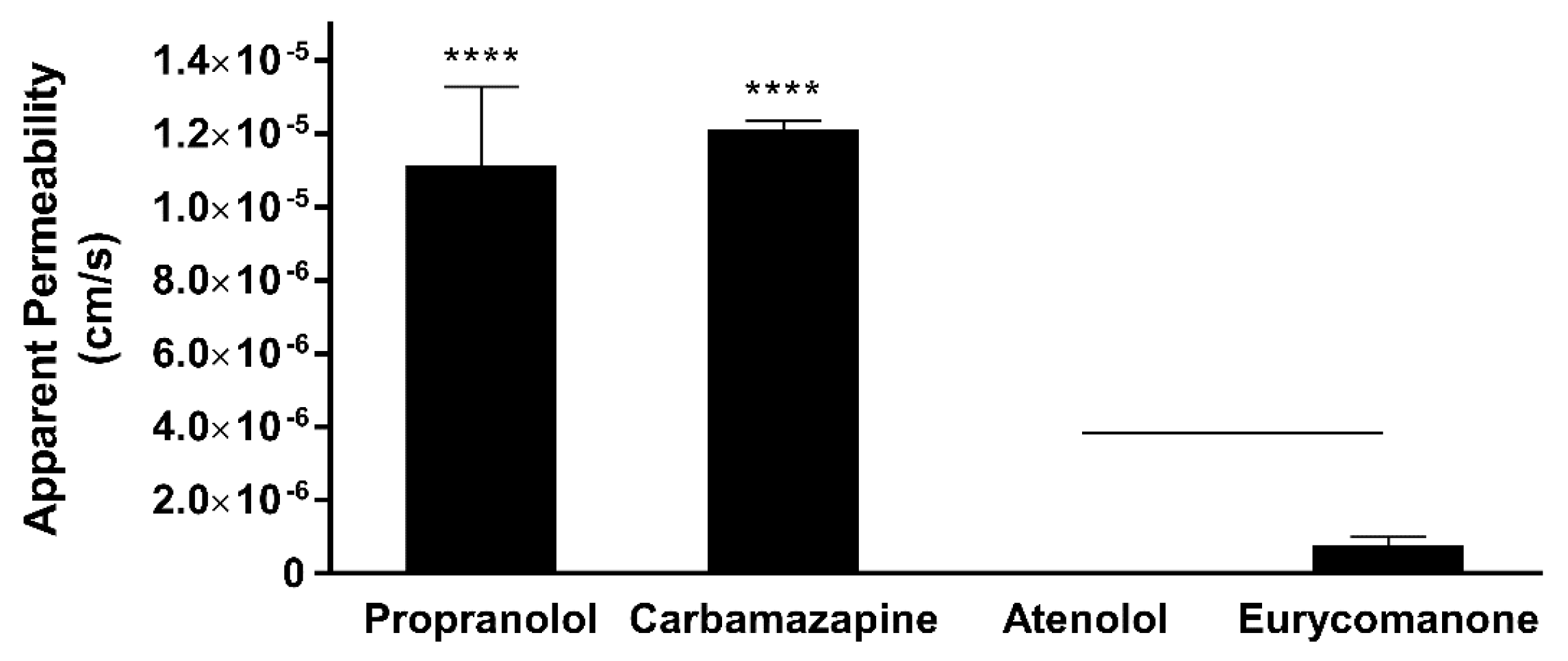
Eurycomanone was added to Pion buffer (Pion Inc., Billerica, MA, USA) pH 7.4 to make a 10 µM working solution. Five microlitres of the lipid mix was applied on the membrane and allowed to dry for 10 min. The working solution (200 µL) was added to the donor plate and Pion buffer (200 µL) were added to the receiver plate. The plate was kept in a moist chamber and left for 18 h at room temperature. The solution from both the donor and receiver plate and the working solution was analysed using LC-MS/MS. This test was carried out in triplicates using propranolol, atenolol, and carbamazepine as standards. The test concentration for eurycomanone is 25 µM.
2.8.2. Caco-2 Cell Permeability Assay
The Caco-2 assay was carried out after seeding the cells onto the membrane to form a confluent monolayer in 21 days. The media is removed from apical (A) and basolateral (B) chambers and the membrane is washed with HBSS buffer. Eurycomanone (10 µM, 200 µL) was added to the A compartment (which represents the intestinal lumen), and buffer (300 µL) was added to the B compartment (representing the blood), and vice versa. The plate was kept in a shaking incubator (37 °C) for 1 h. The amount of eurycomanone that had permeated across the cells was measured by LC-MS/MS, and the apparent permeability (Papp) values and efflux ratio were calculated. The expression of P-glycoprotein (P-gp) by differentiated Caco-2 cells was confirmed by measuring the Papp value and efflux ratio of vinblastine with and without the presence of a competing P-gp substrate, cyclosporine A. Eurycomanone was also measured with the presence of cyclosporine A to investigate whether eurycomanone may be transported via P-gp transporters.
2.9. Liver Microsome Metabolic Stability Assay
The liver microsome stability assay of eurycomanone was estimated using phase I and phase II enzymes in five types of microsomes (mice, rat, dog, monkey, and human). Eurycomanone at 1 mM (20 µL), microsomes at 0.3 mg/mL (20 µL), and 10 mg/mL of alamethicin in 1:1 DMSO and methanol (20 µL) were preincubated at 37 °C for 10 min. The cofactor, 5 mM UDPGA (20 µL), and 1 mM NADPH (20 µL) were added to make the final assay volume of 100 µL. At timepoints 0, 15, 30, 45, 60, and 90 min, 100 µL of the reaction mixture is taken out and added to the stop solution. The supernatants (10,000 rpm, 10 min, 4 °C) were collected and analysed using LC-MS/MS. The standards used for mice and rat liver microsomes were propranolol and dasatinib. The standards used with the dog and monkey liver microsomes were propranolol and midazolam, while the standards used with the human liver microsomes were propranolol and terfinadine.
2.10. Plasma Stability
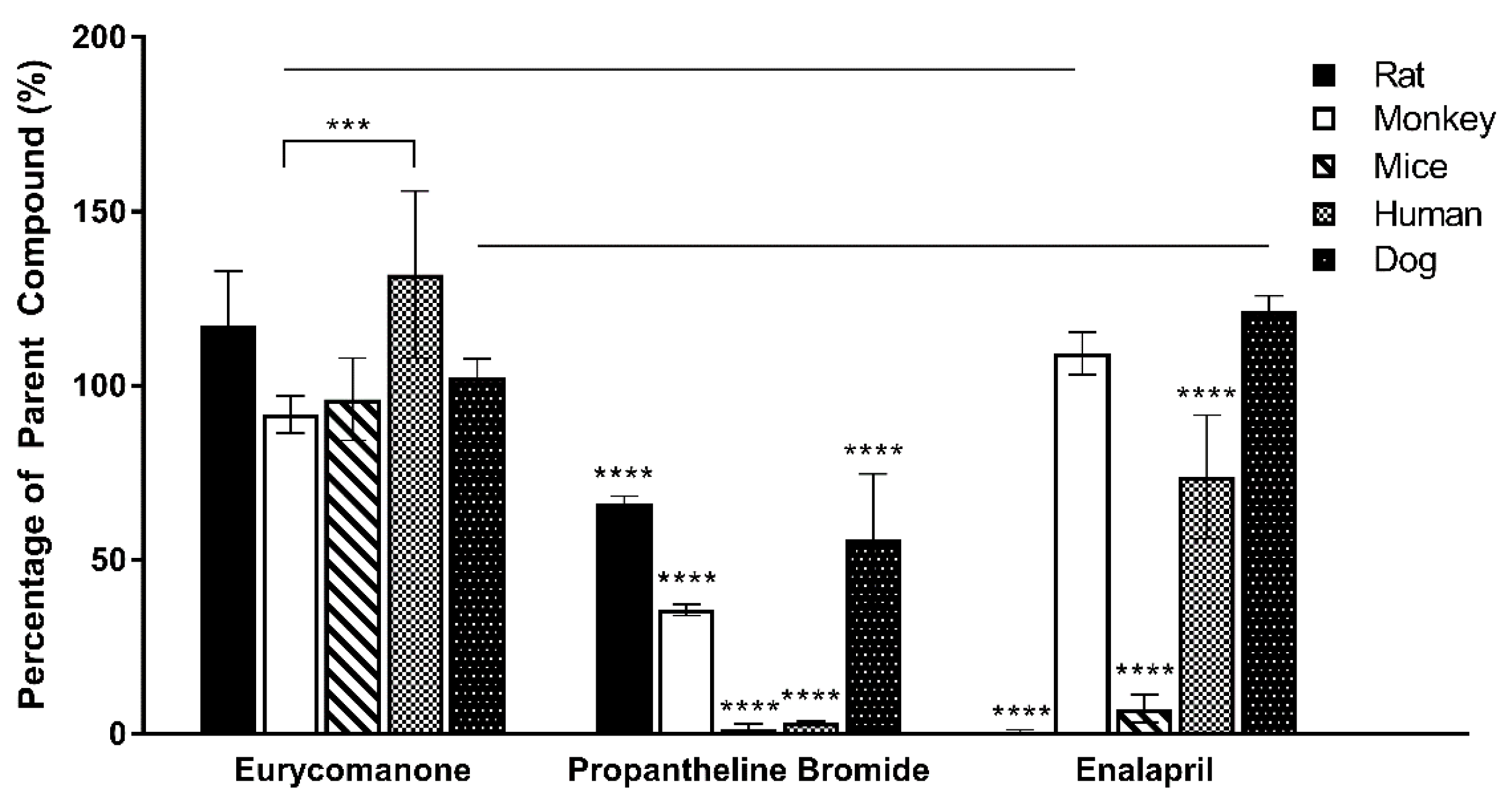
The plasma stability was estimated by preparing 5 µM of eurycomanone in 600 µL plasma in triplicates. The plasma stability of eurycomanone was determined in five types of plasma (mice, rat, dog, monkey, and human). The reaction was kept at 37 °C and ACN (100 µL) was added at timepoints 0, 30 min, and 1, 2, and 4 h to the reaction mixture (100 µL). The supernatant (10,000 rpm, 10 min, 4 °C) was collected and analysed by LC-MS/MS. The standards (propantheline bromide and enalapril) were analysed in triplicates.
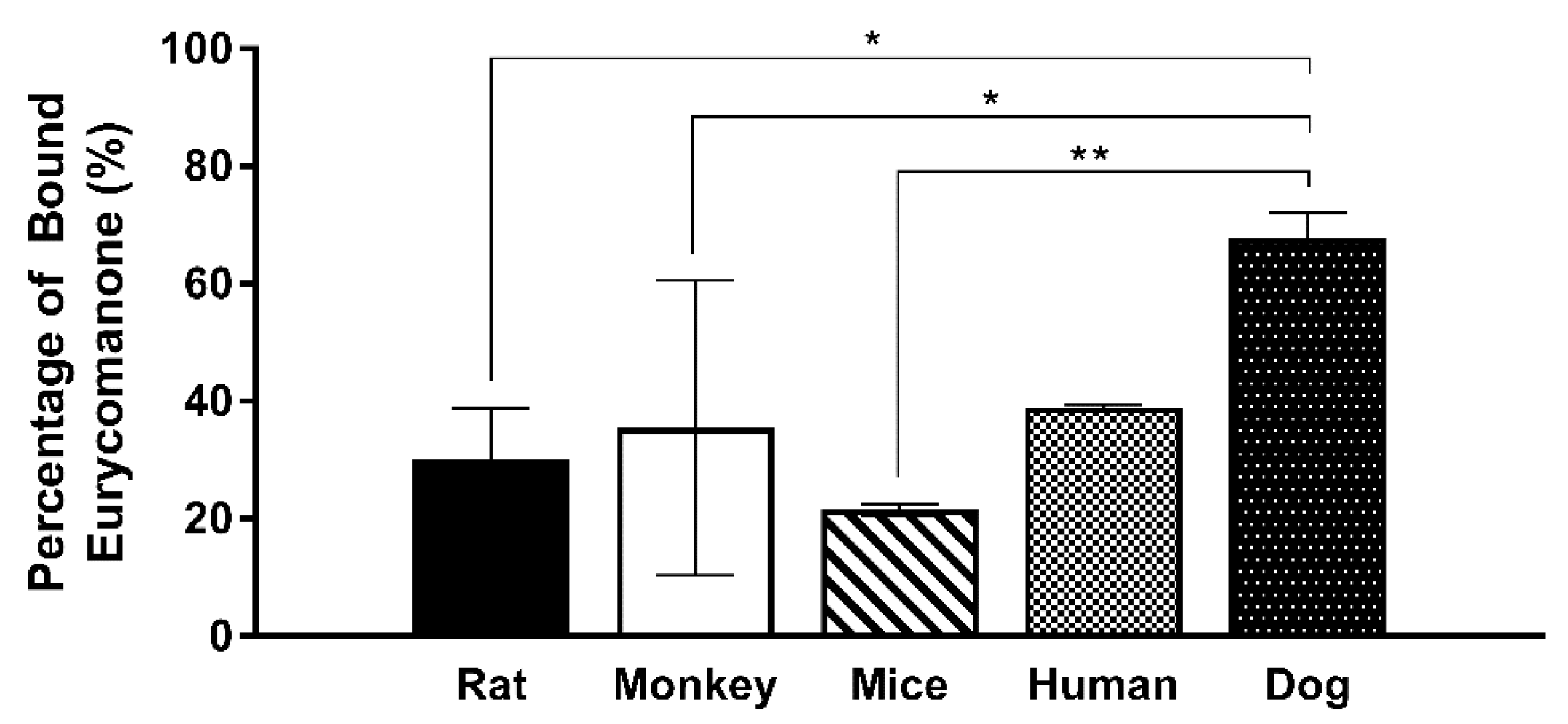
2.11. Plasma Protein Binding Assay
The ability of eurycomanone to bind plasma protein was analysed in five types of plasma (mice, rat, dog, monkey, and human). Each type of plasma was spiked with eurycomanone (1 mM) to make the 10 µM test concentration of eurycomanone in the plasma. Equilibrium dialysis buffer (500 µL) at pH 7.4 was added to the right chamber in rapid equilibrium dialysis (RED), while the spiked plasma (300 µL) was added to the left chamber. The Teflon base plate was covered with aluminium foil and left on the shaking incubator at 37 °C for 4 h. The matrix was balanced by mixing 100 µL of the equilibrium buffer with the spiked plasma from the left chamber in RED before adding the stop solution (350 µL) to precipitate out the protein. The supernatant (10,000 rpm, 10 min, 4 °C) was collected and analysed using LC-MS/MS. The standards (propranolol, warfarin, and acebutalol) were analysed in duplicates.
2.12. Statistical Analysis
The statistical analysis was performed using one-way or two-way ANOVA with either Dunnett or Tukey analysis chosen as the post-hoc analysis (GraphPad Prism, version 7.00 for Windows, GraphPad Software, La Jolla, CA, USA). Statistical significant differences were recognised at p ≤ 0.05.
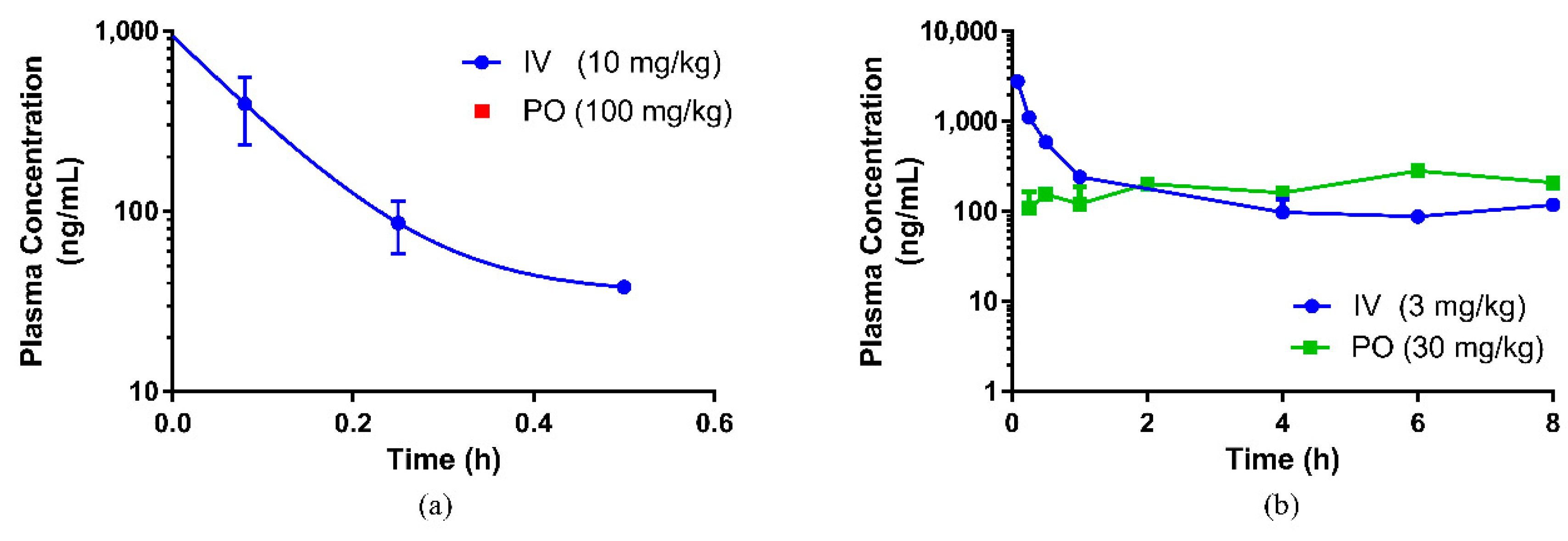
4. Discussion
Eurycomanone has been evaluated for its biological activities via various in vitro methods [
17,
20,
24,
25,
26,
27], while the extract form was mainly used in various in vivo tests [
28,
29,
30,
31,
32]. From this study, the concentration of the pure eurycomanone in rat plasma was at its maximum (C
max 238.3 ng/mL) at 2 h post oral administration, and the bioavailability for the eurycomanone compound was low (11.8%). The findings in this study with regard to eurycomanone’s bioavailability was similar to findings by Low et al. [
22], where it was reported that the bioavailability of the orally administered fortified eurycomanone in
E. longifolia extract was 10.5% and the C
max was found to be 330 ng/mL. There was no notable difference in terms of eurycomanone’s bioavailability and maximum concentration when using a pure compound as in the current study and when compared to using a fortified extract as used by Low et al. [
22]. Rehman and his coworkers, in their investigation, found the C
max to be lower at 40.43 ng/mL for the pure eurycomanone and 9.90 ng/mL for eurycomanone in
E. longifolia extract [
7]. In this study, interspecies variation in the bioavailability of eurycomanone was noted. The pure compound was found to be more bioavailable (higher absorption) in mice (54.9%) than in rats, and the C
max (335 ng/mL) in mice was slightly higher than the levels found in rats. This also indicates that eurycomanone may behave differently in different species and the PK values may not be able to be directly extrapolated from rodents to humans. This species-dependent characteristic of eurycomanone behaviour in rodents may be due to species differences in the metabolism and disposition of eurycomanone, as there was a variation in the absorption, distribution, and metabolism of eurycomanone, in which they were faster in mice than in rats. Two peaks were observed in the PK profiles in both the oral and intravenous profiles. The presence of these secondary peaks may indicate the occurrence of enterohepatic circulation of eurycomanone [
33]; however, this must be further investigated in a separate study. Ma et al. suggest that the secondary peak of eurycomanone may be caused by eurycomanone’s activity as a muscle relaxant, which caused delayed gastric emptying and hence delayed intestinal absorption, resulting in the secondary peak in the profile [
23]. In the present study, the plasma concentration–time profile was only conducted for 8 h, which limits the window of analysis to confirm the second peak.
Previously, Zakaria et al. reported eurycomanone activity in vivo, where eurycomanone was orally administered at 6 mg/kg and 17 mg/kg to mice. At the dose of 17 mg/kg, tumour suppression ability was demonstrated in nude mice with HepG2 cell-induced tumours, which showed that there was sufficient or better bioavailability to exert the reported efficacy [
32]. However, in relation to previous in vitro studies, the C
max value obtained in the current study was much lower than those shown in previous studies of eurycomanone and
E. longifolia extracts (in the range of micrograms to miligrams) [
17,
20,
24,
25,
26,
27]. The reported efficacy via in vitro models may not accurately represent eurycomanone’s level in vivo. Stability in plasma was measured via predetermined time incubation (0 and 30 min and 1, 2, 4 h) of eurycomanone and it was found to be highly stable; nevertheless, eurycomanone’s ability to bind to proteins present in the plasma may affect its availability to its target site of action. However, the plasma protein binding in rats indicates that a low percentage of eurycomanone (30%) is bound to plasma proteins. In other words, the remaining 70% unbound fraction of eurycomanone is freely available and can possibly access the target sites [
34].
The low absorption of eurycomanone in vivo correlates with findings from in vitro studies, namely the lipophilicity, solubility, and permeability assessment of eurycomanone. The lipophilicity of eurycomanone was very low and its solubility was high, which influences its permeation rate via lipid membranes as well as cell membranes such as of Caco-2 cells. The low log D value determined in this study indicates the extent to which eurycomanone was likely to stay in the aqueous environment instead of dissociating into the hydrophobic octanol. This relates to eurycomanone being highly soluble and its low permeation when assayed using the PAMPA method. When eurycomanone’s permeability was assessed using the Caco-2 cells, the results also indicated that a low amount of eurycomanone permeated the cell monolayer. However, this may be due to the condition where eurycomanone, which was not lipophilic enough to be absorbed, accumulates on the cell monolayer, introducing a sink condition [
35]. This accumulation then draws the compound to cross the membrane, which may not be due to the compound’s innate ability to cross the membrane freely.
The ability of the compound to cross the biological membrane without the use of transporters is possible when the compound of interest is in its uncharged state. Eurycomanone is predicted to have an acid dissociation constant (pK
a) value of 11 [
36]. The pH values of the environments in the stomach and small intestine vary in the different sections and depending on the fasted or fed state. The pH range in the stomach in its fasted state is between 1.4 and 2.1; while in its fed state, it is in between 4.3 and 5.4. In the small intestine, the pH values of the different segments in their fasted state are 4.9–6.4 (duodenum); 4.4–6.6 (jejunum), and 6.5–7.4 (ileum); while in their fed state, the pH values are 4.2–6.1 (duodenum); 5.2–6.2 (jejunum), and 6.8–7.5 (ileum) [
37]. The changing pH values then affect the ionic state of eurycomanone. The predicted pK
a for eurycomanone was high, which means that in a lower pH environment, eurycomanone exists in its ionised form. This then prevents eurycomanone from crossing the membrane barriers passively.
As determined in the Caco-2 permeability assessment, eurycomanone was not an active transporter P-gp substrate. The chemical stability assay conducted using the fasted and fed state simulative buffer system indicates that eurycomanone was stable; however, its ionic state could not be confirmed. The plasma stability and liver metabolic stability study also showed eurycomanone to be stable in the different matrices, indicating that eurycomanone may stay intact and survive first-pass metabolism in vivo. This study hypothesised that when eurycomanone was administered orally, only a small amount crosses the membrane to enter the blood stream, while when it was administered intravenously, only a small amount of eurycomanone permeates into tissues, while most remained in the blood in its ionised form. The small amount of eurycomanone which may have been absorbed (as a low amount was detected in liver tissues) could still potentially exert the desired effects of eurycomanone or E. longifolia extract in general.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}