In Vitro, Ex Vivo and In Vivo Evaluation of Microcontainers for Oral Delivery of Insulin

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Microcontainer Fabrication, Characterization, Drug Loading and Polymer Coating
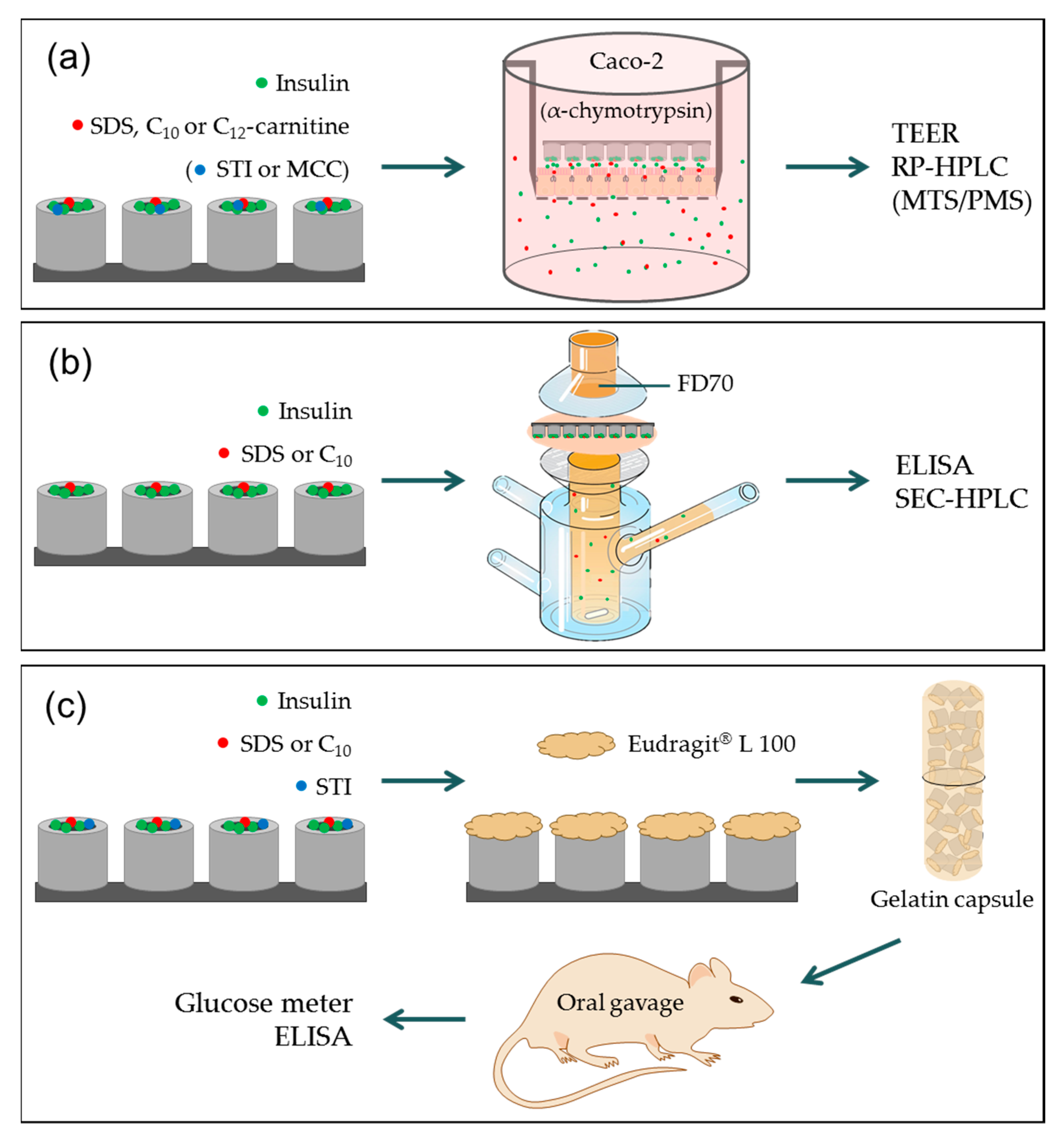
2.3. In Vitro Permeation Studies
2.4. Combined In Vitro Permeation and Proteolysis
2.5. Ex Vivo Intestinal Permeation Studies
2.6. In Vivo Studies
2.7. HPLC Quantification of Insulin and FD70
2.8. Data Analysis
3. Results and Discussion
3.1. Microcontainer Fabrication and Characterization
3.2. In Vitro Permeation Studies
3.3. Ex Vivo Intestinal Permeation Studies
3.4. Combined In Vitro Permeation and Proteolysis
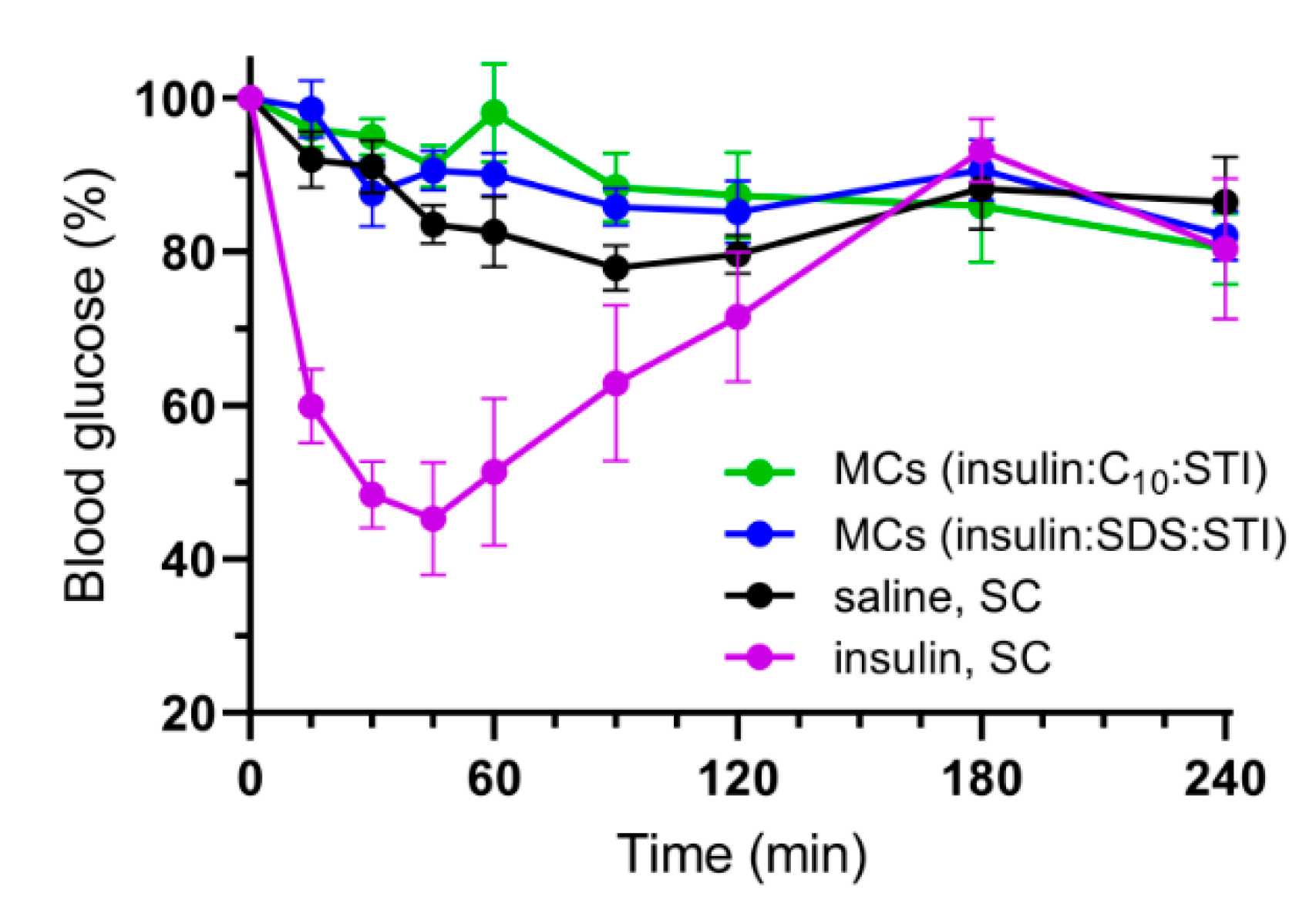
3.5. In Vivo Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ismail, R.; Csóka, I. Novel strategies in the oral delivery of antidiabetic peptide drugs–Insulin, GLP 1 and its analogs. Eur. J. Pharm. Biopharm. 2017, 115, 257–267. [Google Scholar] [CrossRef]
- Moroz, E.; Matoori, S.; Leroux, J.-C. Oral delivery of macromolecular drugs: Where we are after almost 100 years of attempts. Adv. Drug Deliv. Rev. 2016, 101, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Abramson, A.; Caffarel-Salvador, E.; Khang, M.; Dellal, D.; Silverstein, D.; Gao, Y.; Frederiksen, M.R.; Vegge, A.; Hubálek, F.; Water, J.J.; et al. An ingestible self-orienting system for oral delivery of macromolecules. Science 2019, 363, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, T.A.S.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Bernkop-Schnürch, A. The use of inhibitory agents to overcome the enzymatic barrier to perorally administered therapeutic peptides and proteins. J. Control. Release 1998, 52, 1–16. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Buckley, S.T.; Bækdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Rønne, J.; Madsen, K.G.; Schéele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018, 10, eaar7047. [Google Scholar] [CrossRef]
- Rembratt, A.; Graugaard-Jensen, C.; Senderovitz, T.; Norgaard, J.P.; Djurhuus, J.C. Pharmacokinetics and pharmacodynamics of desmopressin administered orally versus intravenously at daytime versus night-time in healthy men aged 55–70 years. Eur. J. Clin. Pharmacol. 2004, 60, 397–402. [Google Scholar] [CrossRef]
- Halberg, I.B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Mörschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: A randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179–188. [Google Scholar] [CrossRef]
- Maher, S.; Leonard, T.W.; Jacobsen, J.; Brayden, D.J. Safety and efficacy of sodium caprate in promoting oral drug absorption: From in vitro to the clinic. Adv. Drug Deliv. Rev. 2009, 61, 1427–1449. [Google Scholar] [CrossRef]
- Banerjee, A.; Lee, J.; Mitragotri, S. Intestinal mucoadhesive devices for oral delivery of insulin. Bioeng. Transl. Med. 2016, 1, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, J.R.; Jepsen, M.L.; Nielsen, L.H.; Dufva, M.; Nielsen, H.M.; Rades, T.; Boisen, A.; Müllertz, A. Microcontainers for oral insulin delivery—In vitro studies of permeation enhancement. Eur. J. Pharm. Biopharm. 2019, 143, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Chirra, H.D.; Shao, L.; Ciaccio, N.; Fox, C.B.; Wade, J.M.; Ma, A.; Desai, T.A. Planar Microdevices for Enhanced In Vivo Retention and Oral Bioavailability of Poorly Permeable Drugs. Adv. Healthc. Mater. 2014, 3, 1648–1654. [Google Scholar]
- Nielsen, L.H.; Keller, S.S.; Boisen, A. Microfabricated devices for oral drug delivery. Lab Chip 2018, 18, 2348–2358. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, L.H.; Keller, S.S.; Gordon, K.C.; Boisen, A.; Rades, T.; Müllertz, A. Spatial confinement can lead to increased stability of amorphous indomethacin. Eur. J. Pharm. Biopharm. 2012, 81, 418–425. [Google Scholar] [CrossRef]
- Petersen, R.S.; Mahshid, R.; Andersen, N.K.; Keller, S.S.; Hansen, H.N.; Boisen, A. Hot embossing and mechanical punching of biodegradable microcontainers for oral drug delivery. Microelectron. Eng. 2015, 133, 104–109. [Google Scholar] [CrossRef]
- Petersen, R.S.; Keller, S.S.; Boisen, A. Hot punching of high-aspect-ratio 3D polymeric microstructures for drug delivery. Lab Chip 2015, 15, 2576–2579. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, L.H.; Melero, A.; Keller, S.S.; Jacobsen, J.; Garrigues, T.; Rades, T.; Müllertz, A.; Boisen, A. Polymeric microcontainers improve oral bioavailability of furosemide. Int. J. Pharm. 2016, 504, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Dalskov Mosgaard, M.; Strindberg, S.; Abid, Z.; Singh Petersen, R.; Højlund Eklund Thamdrup, L.; Joukainen Andersen, A.; Sylvest Keller, S.; Müllertz, A.; Hagner Nielsen, L.; Boisen, A. Ex vivo intestinal perfusion model for investigating mucoadhesion of microcontainers. Int. J. Pharm. 2019, 570, 118658. [Google Scholar] [CrossRef]
- Lindmark, T.; Schipper, N.; Lazorová, L.; Boer, A.G.D.; Artursson, P. Absorption Enhancement in Intestinal Epithelial Caco-2 Monolayers by Sodium Caprate: Assessment of Molecular Weight Dependence and Demonstration of Transport Routes. J. Drug Target. 1998, 5, 215–223. [Google Scholar] [CrossRef]
- Anderberg, E.K.; Artursson, P. Epithelial Transport of Drugs in Cell Culture. VIII: Effects of Sodium Dodecyl Sulfate on Cell Membrane and Tight Junction Permeability in Human Intestinal Epithelial (Caco-2) Cells. J. Pharm. Sci. 1993, 82, 392–398. [Google Scholar] [CrossRef]
- Brayden, D.J.; Maher, S.; Bahar, B.; Walsh, E. Sodium caprate-induced increases in intestinal permeability and epithelial damage are prevented by misoprostol. Eur. J. Pharm. Biopharm. 2015, 94, 194–206. [Google Scholar] [CrossRef]
- Mazzoni, C.; Jacobsen, R.D.; Mortensen, J.; Jørgensen, J.R.; Vaut, L.; Jacobsen, J.; Gundlach, C.; Müllertz, A.; Nielsen, L.H.; Boisen, A. Polymeric Lids for Microcontainers for Oral Protein Delivery. Macromol. Biosci. 2019, 19, 1900004. [Google Scholar] [CrossRef] [Green Version]
- Antunes, F.; Andrade, F.; Ferreira, D.; Nielsen, H.M.; Sarmento, B. Models to predict intestinal absorption of therapeutic peptides and proteins. Curr. Drug Metab. 2013, 14, 4–20. [Google Scholar] [CrossRef]
- Pegg, D.E. The relevance of ice crystal formation for the cryopreservation of tissues and organs. Cryobiology 2010, 60, S36–S44. [Google Scholar] [CrossRef]
- McGowan, I.; Tanner, K.; Elliott, J.; Ibarrondo, J.; Khanukhova, E.; McDonald, C.; Saunders, T.; Zhou, Y.; Anton, P.A. Nonreproducibility of “Snap-Frozen” Rectal Biopsies for Later Use in Ex Vivo Explant Infectibility Studies. AIDS Res. Hum. Retrovir. 2012, 28, 1509–1512. [Google Scholar] [CrossRef] [Green Version]
- Procedure for Enzymatic Assay of α-Chymotrypsin (EC 3.4.21.1). Available online: https://www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-chymotrypsin.html (accessed on 11 September 2019).
- Bock, U.; Kolac, C.; Borchard, G.; Koch, K.; Fuchs, R.; Streichhan, P.; Lehr, C.M. Transport of proteolytic enzymes across Caco-2 cell monolayers. Pharm. Res. 1998, 15, 1393–1400. [Google Scholar] [CrossRef]
- Analytical Enzyme Chymotrypsin. Available online: https://www.sigmaaldrich.com/life-science/metabolomics/enzyme-explorer/analytical-enzymes/chymotrypsin.html (accessed on 11 September 2019).
- Zhang, L.; Jiang, H.; Zhu, W.; Wu, L.; Song, L.; Wu, Q.; Ren, Y. Improving the Stability of Insulin in Solutions Containing Intestinal Proteases in Vitro. Int. J. Mol. Sci. 2008, 9, 2376–2387. [Google Scholar] [CrossRef]
- Abid, Z.; Strindberg, S.; Javed, M.M.; Mazzoni, C.; Vaut, L.; Nielsen, L.H.; Gundlach, C.; Petersen, R.S.; Müllertz, A.; Boisen, A.; et al. Biodegradable microcontainers–towards real life applications of microfabricated systems for oral drug delivery. Lab Chip 2019, 19, 2905–2914. [Google Scholar] [CrossRef]
- Vaut, L.; Jensen, K.E.; Tosello, G.; Khosla, A.; Furukawa, H.; Boisen, A. Additive Manufacturing of Microreservoir Devices for Oral Drug Delivery Using an Acculas BA-30 Micro-Stereolithography Instrument: A Feasibility Study. J. Electrochem. Soc. 2019, 166, B3257–B3263. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jørgensen, J.R.; Yu, F.; Venkatasubramanian, R.; Nielsen, L.H.; Nielsen, H.M.; Boisen, A.; Rades, T.; Müllertz, A. In Vitro, Ex Vivo and In Vivo Evaluation of Microcontainers for Oral Delivery of Insulin. Pharmaceutics 2020, 12, 48. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12010048
Jørgensen JR, Yu F, Venkatasubramanian R, Nielsen LH, Nielsen HM, Boisen A, Rades T, Müllertz A. In Vitro, Ex Vivo and In Vivo Evaluation of Microcontainers for Oral Delivery of Insulin. Pharmaceutics. 2020; 12(1):48. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12010048
Chicago/Turabian StyleJørgensen, Jacob Rune, Feiyang Yu, Ramakrishnan Venkatasubramanian, Line Hagner Nielsen, Hanne Mørck Nielsen, Anja Boisen, Thomas Rades, and Anette Müllertz. 2020. "In Vitro, Ex Vivo and In Vivo Evaluation of Microcontainers for Oral Delivery of Insulin" Pharmaceutics 12, no. 1: 48. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12010048